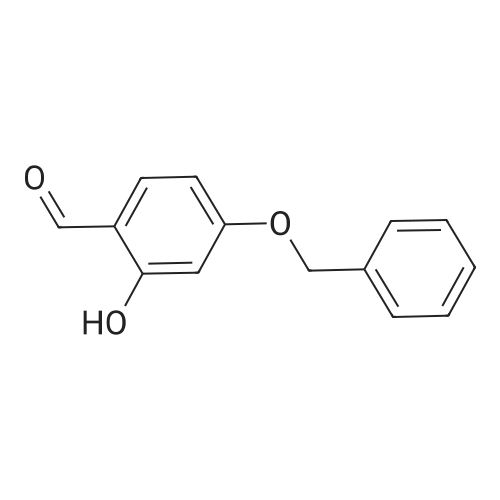
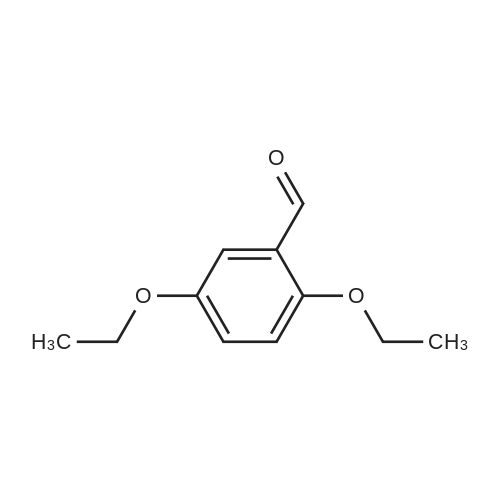
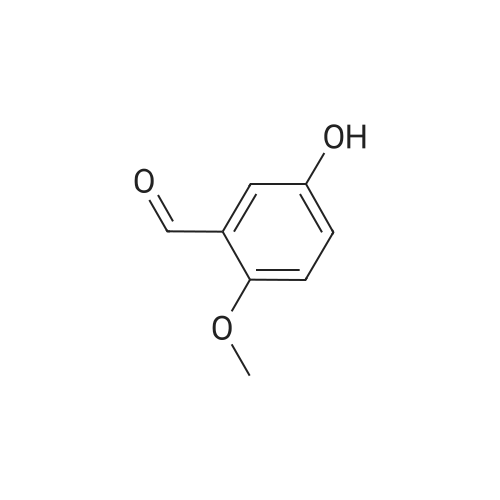
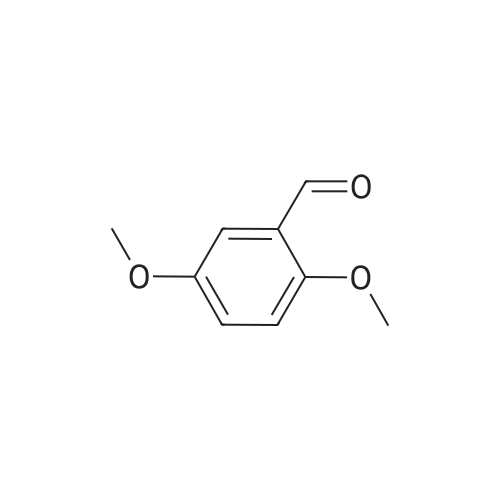
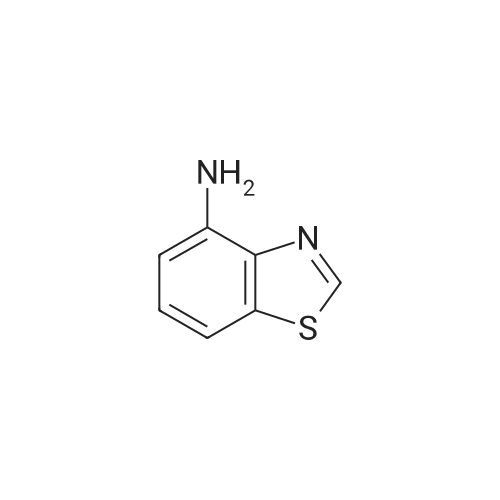
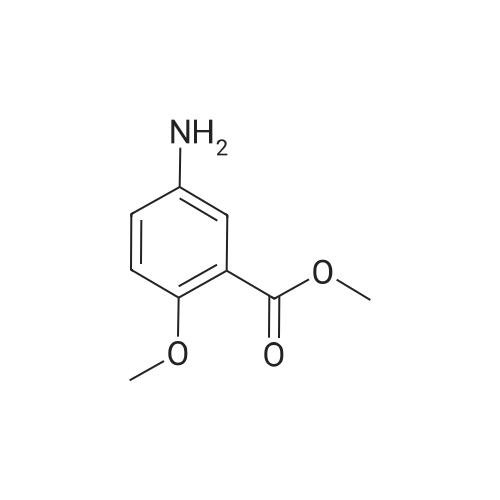
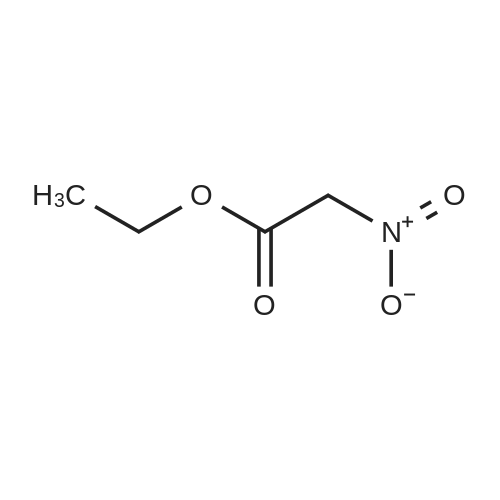
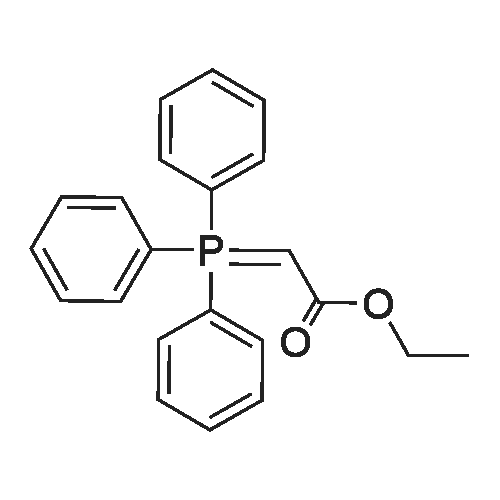
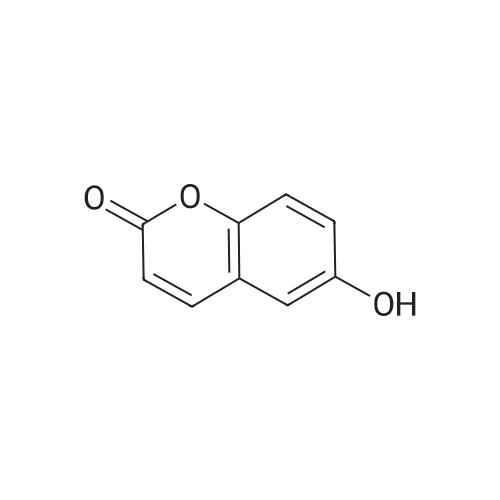
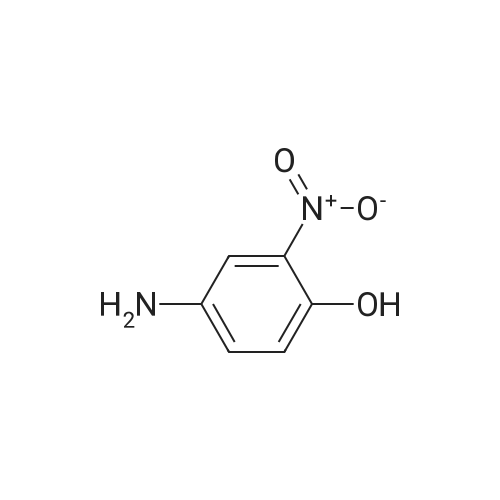
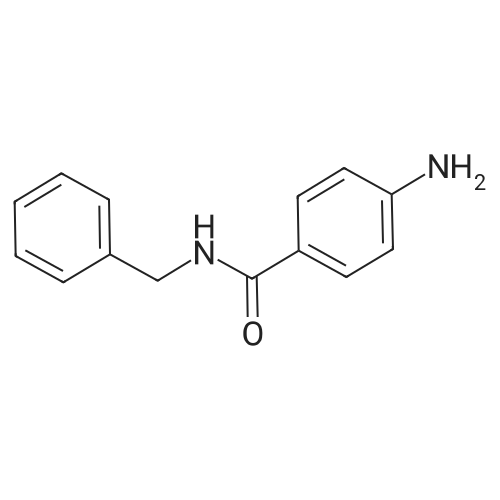
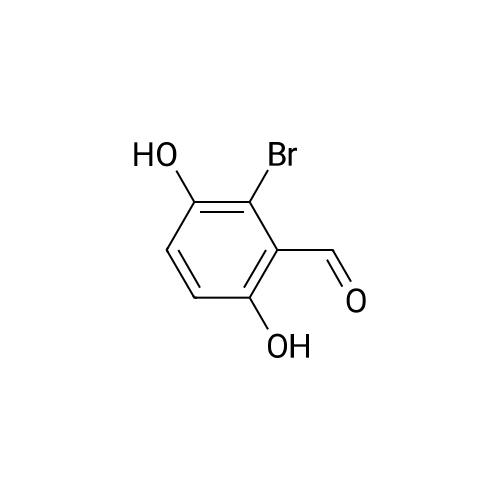
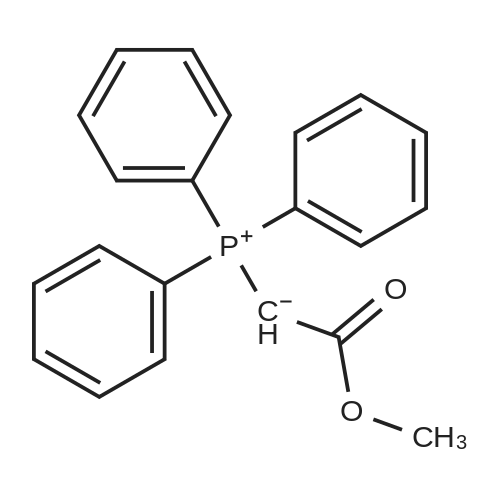
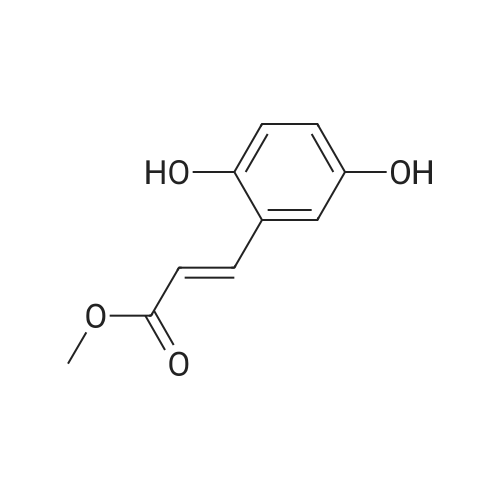
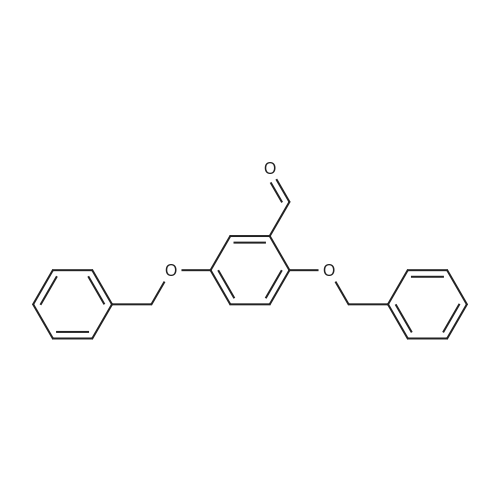
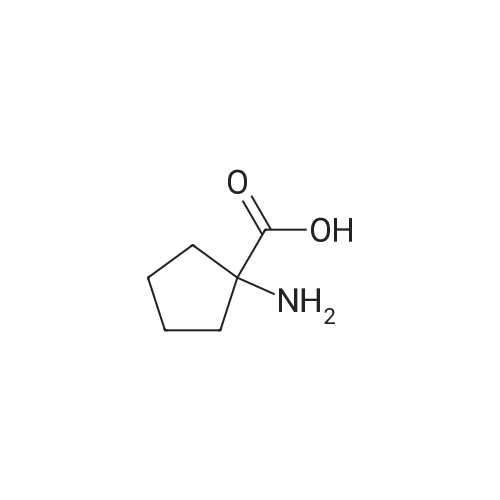
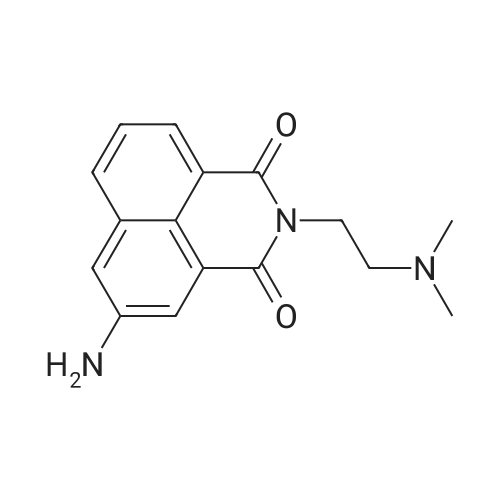
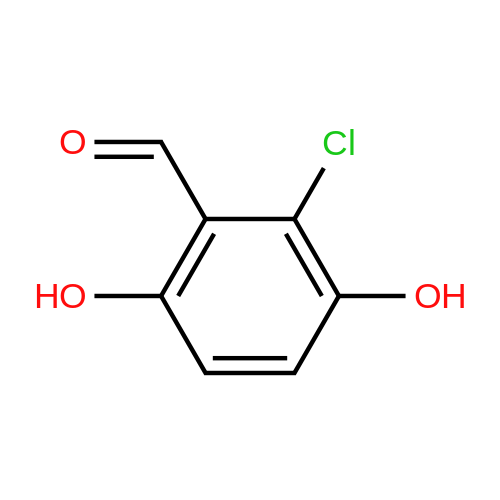
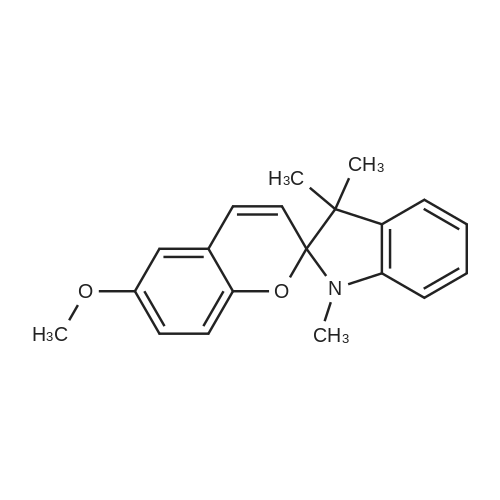
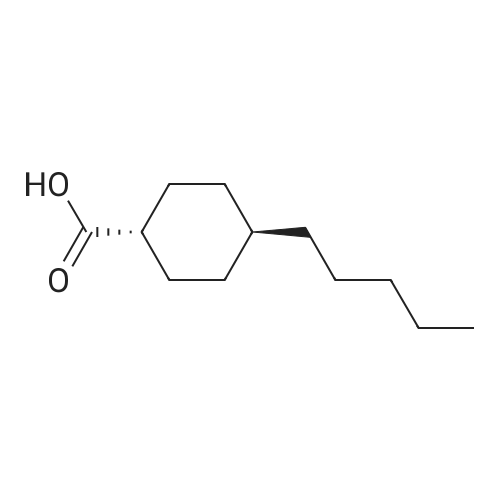
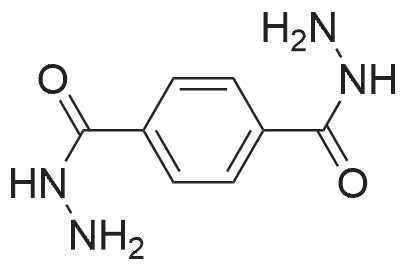
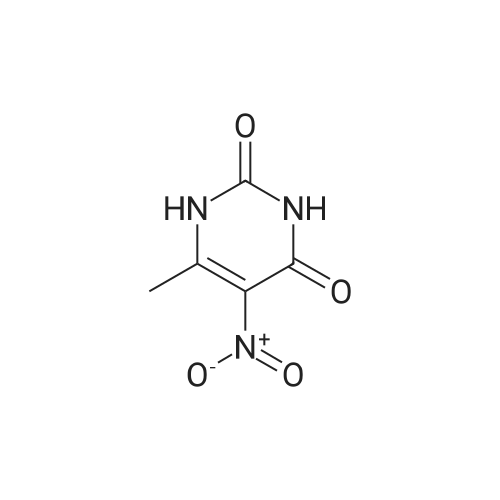
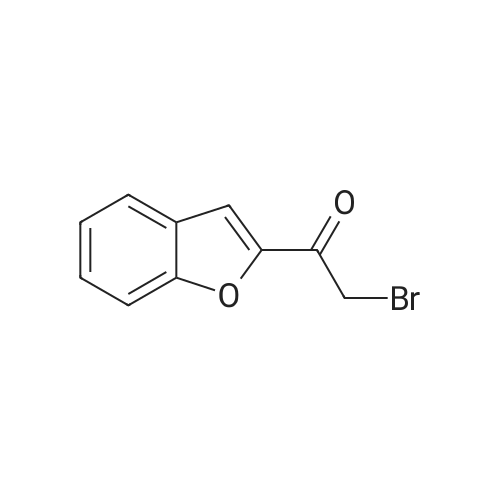
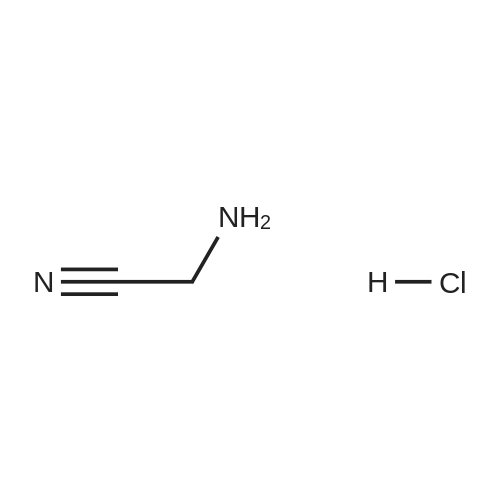
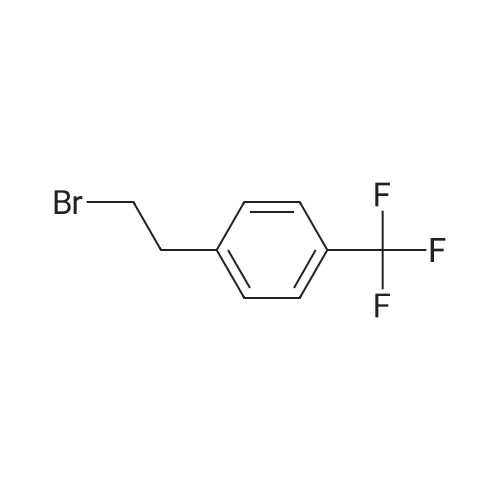
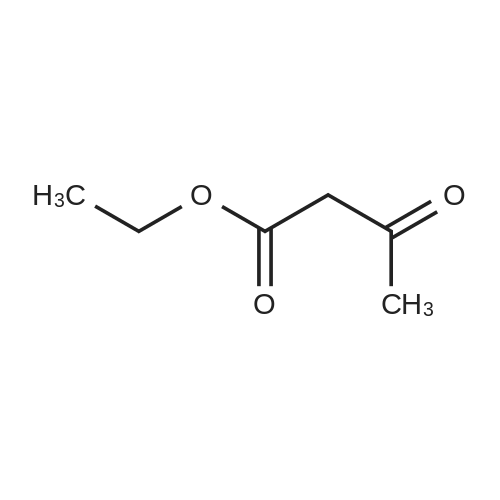
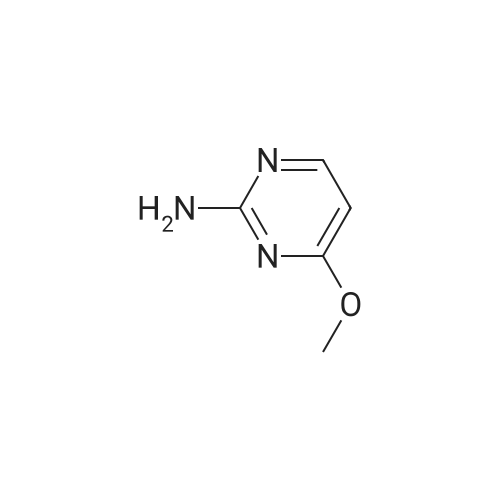
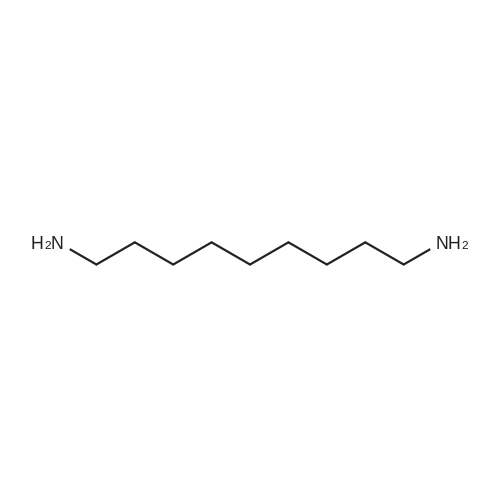
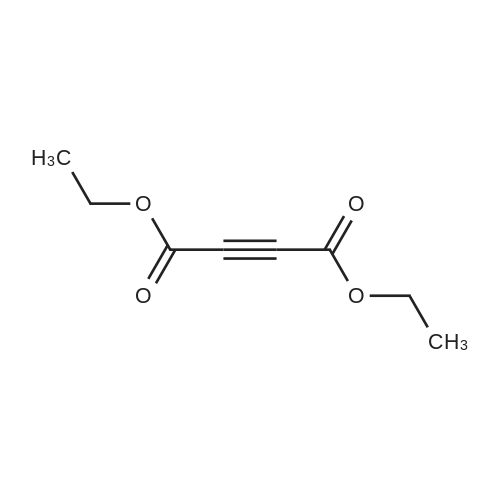
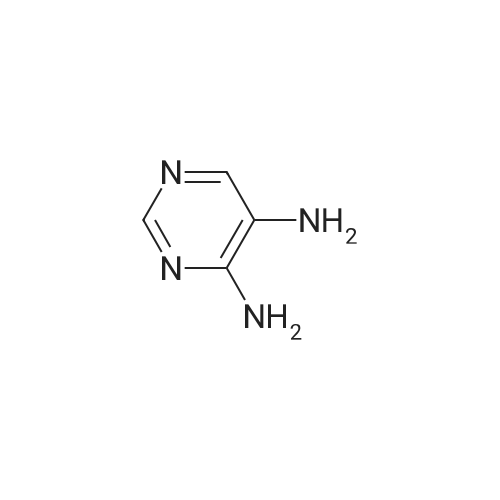
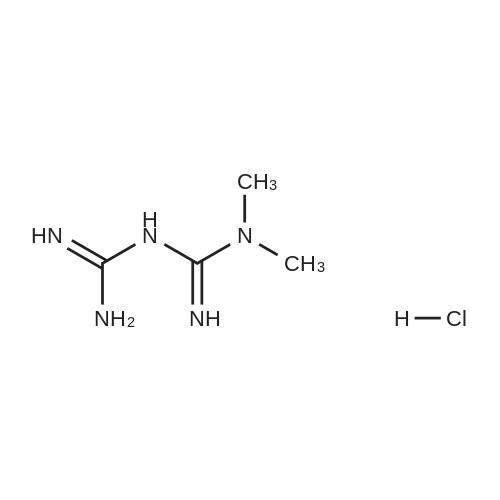
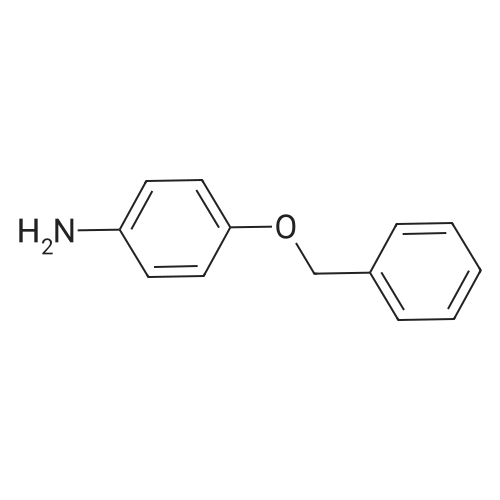
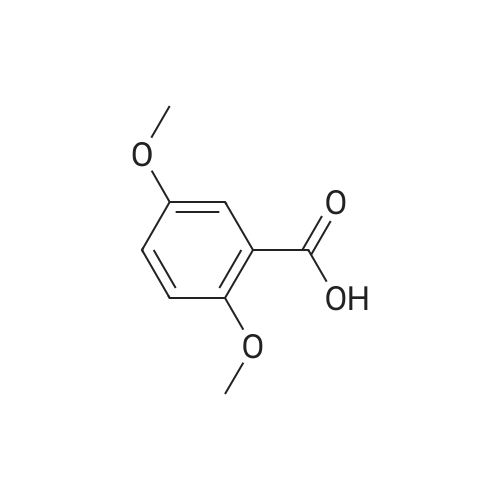
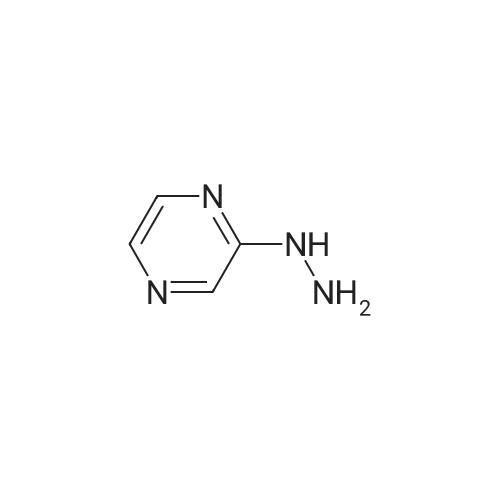
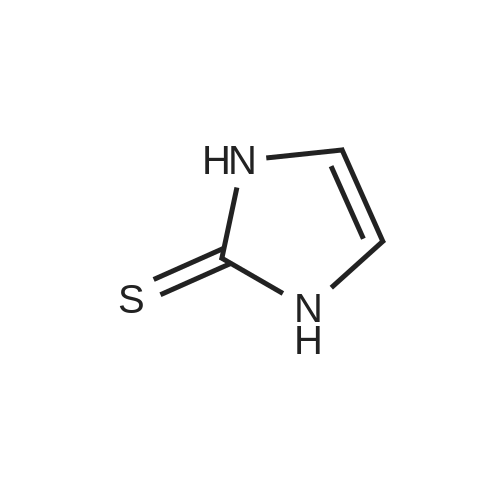
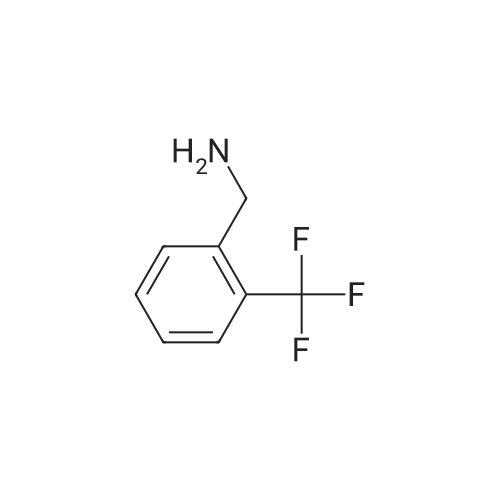
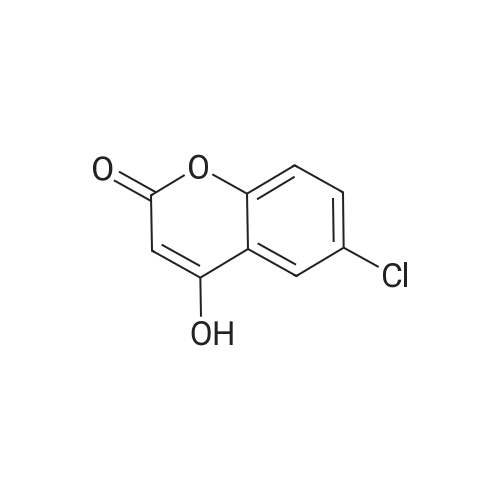
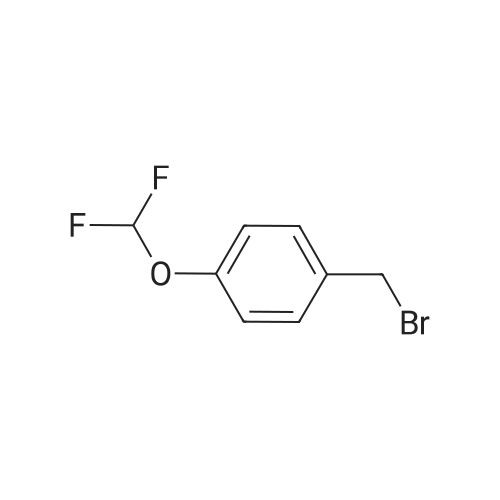
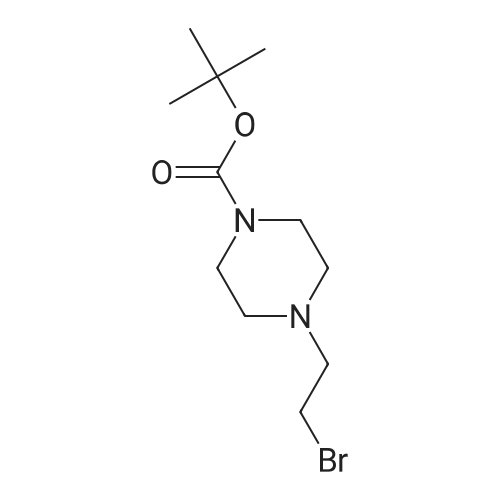
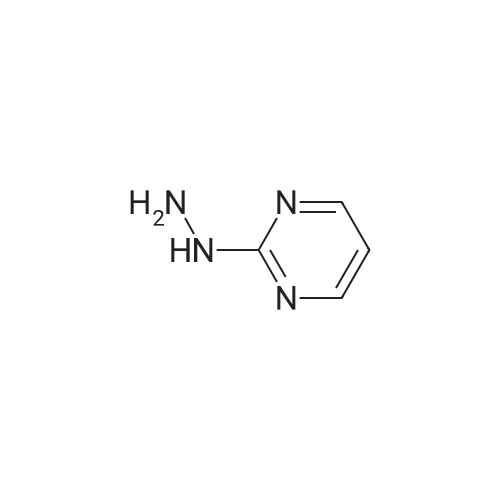
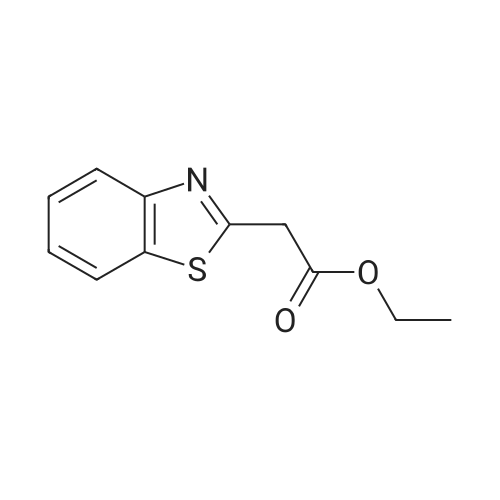
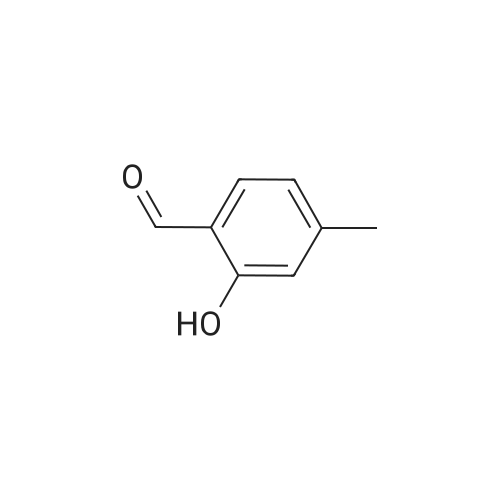
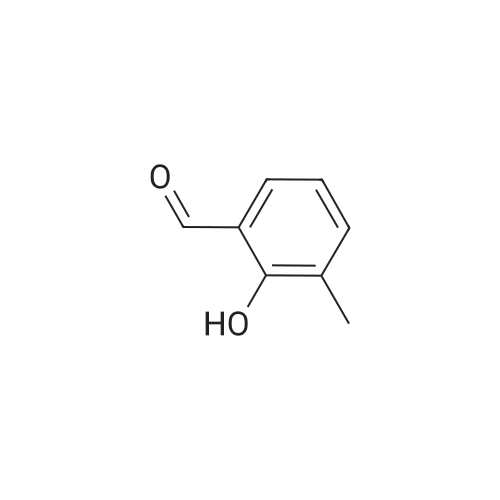
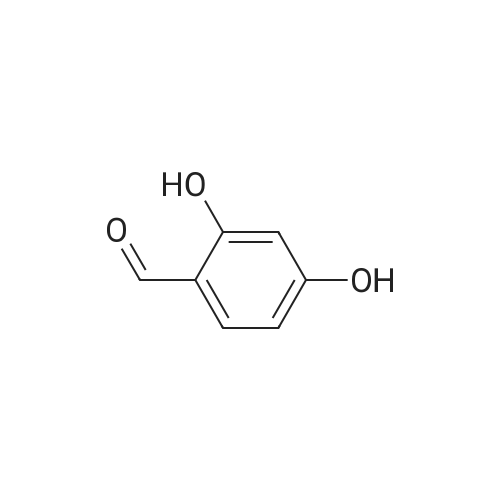
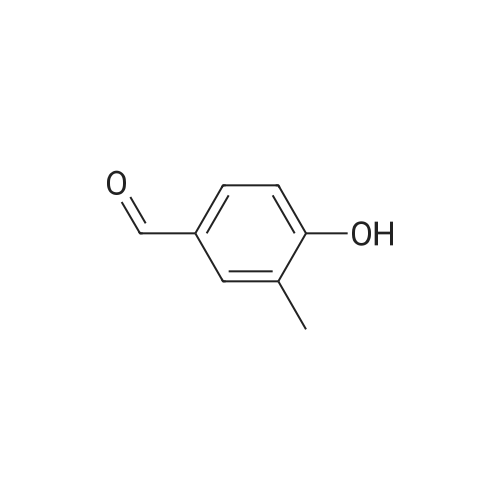
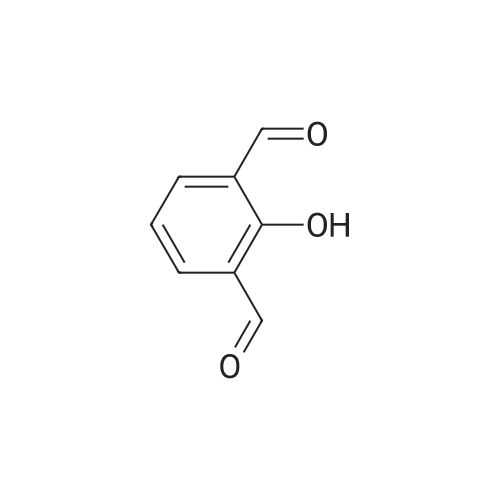
| 7.1 g |
With dmap; diisopropyl-carbodiimide In dichloromethane at 20℃; for 10h; Inert atmosphere; Cooling with ice; |
9 Production of the Compound Represented by Formula (I-109)
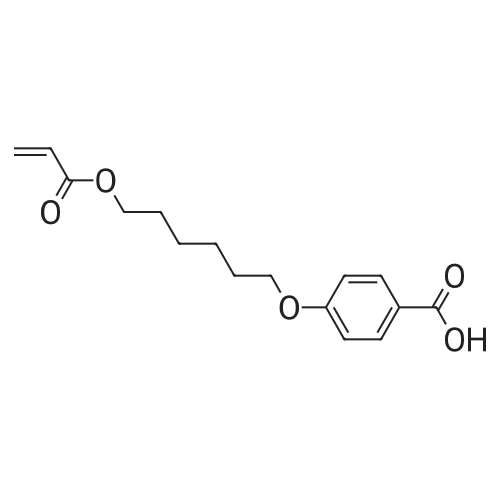
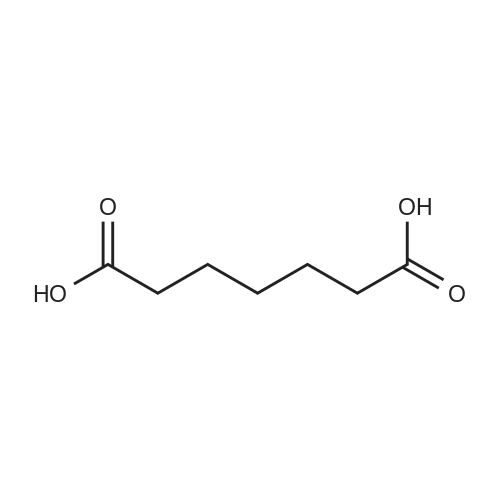
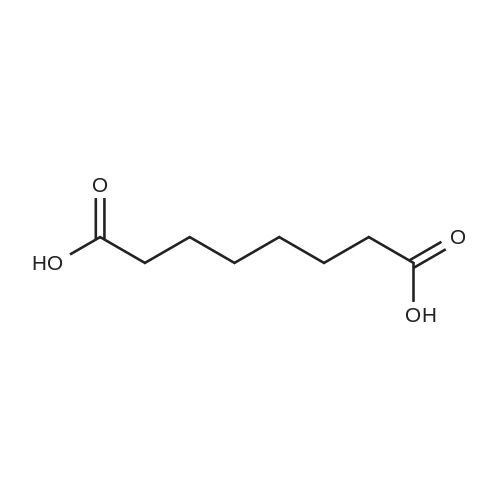
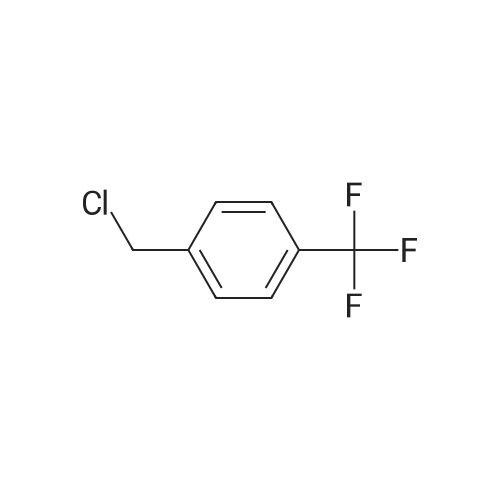
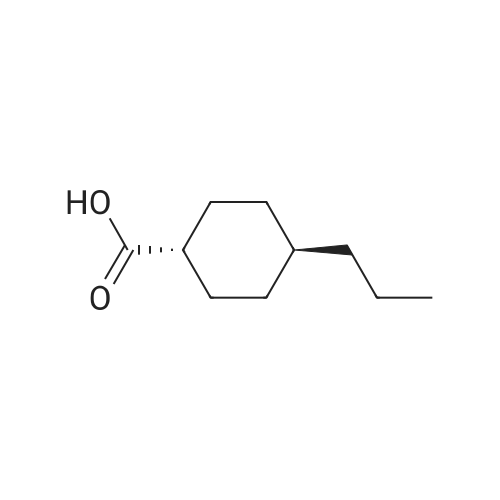
To a reaction container equipped with a Dean and Stark device, 30.0 g of the compound represented by Formula (I-109-1), 19.0 g of acrylic acid, 2.1 g of p-toluenesulfonic acid monohydrate, 300 mL of cyclohexane, and 150 mL of diisopropyl ether were added. The resulting mixture was heated to reflux for 12 hours while water was removed from the mixture. Subsequently, dilution with dichloromethane and washing with a 5%-sodium hydrogencarbonate aqueous solution and then with a saline solution were performed. Then, purification was performed by column chromatography (silica gel, dichloromethane). Hereby, 33.5 g of the compound represented by Formula (I-109-2) was prepared. (0173) To a reaction container, 10.0 g of the compound represented by Formula (I-109-2), 28.9 g of hydroquinone, 21.7 g of potassium carbonate, and 150 mL of acetone were added. The resulting mixture was heated to reflux for eight hours. After the mixture had been poured into 5%-hydrochloric acid, extraction with dichloromethane and cleaning with a saline solution were performed. Then, purification was performed by column chromatography (alumina, dichloromethane) and recrystallization (dichloromethane/hexane). Hereby, 9.7 g of the compound represented by Formula (I-109-3) was prepared. (0174) In a nitrogen atmosphere, 9.7 g of the compound represented by Formula (I-109-3), 7.9 g of the compound represented by Formula (I-109-4), 0.4 g of N,N-dimethylaminopyridine, and 100 mL of dichloromethane were added to a reaction container. While the resulting mixture was cooled with ice, 5.6 g of diisopropylcarbodiimide was added dropwise to the mixture, which was then stirred at room temperature for 6 hours. After the precipitate had been filtered away, the filtrate was washed with 1%-hydrochloric acid, with water, and then with a saline solution. Subsequently, purification was performed by column chromatography (alumina, dichloromethane) and recrystallization (dichloromethane/methanol). Hereby, 11.9 g of the compound represented by Formula (I-109-5) was prepared. (0175) To a reaction container, 11.9 g of the compound represented by Formula (I-109-5) and 80 mL of dichloromethane were added. To the resulting mixture, 20 mL of trifluoroacetic acid was added dropwise. The mixture was then stirred for eight hours. After the solvent had been distilled away, diisopropyl ether was added to the mixture to precipitate a solid, which was then filtered. The solid was washed with diisopropyl ether and subsequently dried. Hereby, 10.7 g of the compound represented by Formula (I-109-6) was prepared. (0176) In a nitrogen atmosphere, 9.1 g of the compound represented by Formula (I-109-6), 1.5 g of the compound represented by Formula (I-109-7), 0.1 g of N,N-dimethylaminopyridine, and 150 mL of dichloromethane were added to a reaction container. While the resulting mixture was cooled with ice, 3.4 g of diisopropylcarbodiimide was added dropwise to the mixture, which was then stirred at room temperature for 10 hours. After the precipitate had been filtered away, the filtrate was washed with 1%-hydrochloric acid, with water, and then with a saline solution. After recrystallization (dichloromethane/methanol) had been performed, purification was performed by column chromatography (silica gel, dichloromethane) and recrystallization (dichloromethane/methanol). Hereby, 7.1 g of the compound represented by Formula (I-109-8) was prepared. (0177) To a reaction container, 10.0 g of the compound represented by Formula (I-109-9), 13.8 g of the compound represented by Formula (I-109-2), 12.5 g of potassium carbonate, and 100 mL of N,N-dimethylformamide were added. The resulting mixture was stirred for 8 hours while being heated at 70° C. After dilution had been performed with dichloromethane, washing with water and then with a saline solution was performed. Subsequently, purification was performed by column chromatography (alumina, dichloromethane). Hereby, 11.6 g of the compound represented by Formula (I-109-10) was prepared. (0178) To a reaction container, 2.0 g of the compound represented by Formula (I-109-10), 5.9 g of the compound represented by Formula (I-109-8), 0.7 g of (±)-10-camphorsulfonic acid, 24 mL of tetrahydrofuran, and 24 mL of ethanol were added. The resulting mixture was stirred for 8 hours while being heated at 50° C. After the solvent had been distilled away, purification was performed by column chromatography (silica gel, dichloromethane) and recrystallization (dichloromethane/methanol). Hereby, 5.4 g of the compound represented by Formula (I-109) was prepared. (0179) 1H NMR (CDCl3) δ 1.24 (m, 4H), 1.48-1.93 (m, 28H), 2.08 (t, 4H), 2.23 (m, 4H), 2.54 (m, 4H), 3.94 (t, 4H), 4.17 (t, 4H), 4.53 (t, 2H), 4.65 (t, 2H), 5.82 (dd, 3H), 6.12 (dd, 3H), 6.40 (dd, 3H), 6.88 (m, 6H), 6.97 (dd, 4H), 7.16 (t, 1H), 7.34 (t, 1H), 7.54 (d, 1H), 7.66 (d, 1H), 7.70 (d, 1H), 8.36 (s, 1H) ppm. (0180) LCMS: 1240 [M+1] |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping