
Abstract
In the last 20 years, direct alcohol fuel cells (DAFCs) have been the subject of tremendous research efforts for the potential application as on-demand power sources. Two leading technologies respectively based on proton exchange membranes (PEMs) and anion exchange membranes (AEMs) have emerged: the first one operating in an acidic environment and conducting protons; the second one operating in alkaline electrolytes and conducting hydroxyl ions. In this review, we present an analysis of the state-of-the-art acidic and alkaline DAFCs fed with methanol and ethanol with the purpose to support a comparative analysis of acidic and alkaline systems, which is missing in the current literature. A special focus is placed on the effect of the reaction stoichiometry in acidic and alkaline systems. Particularly, we point out that, in alkaline systems, OH− participates stoichiometrically to reactions, and that alcohol oxidation products are anions. This aspect must be considered when designing the fuel and when making an energy evaluation from a whole system perspective.
Graphical Abstract

Similar content being viewed by others

1 Introduction
Fuel cells (FCs) are an electrochemical energy technology that directly convert the chemical energy of a fuel into electrical energy [1]. Such energy conversion is based on the concurrent electrochemical oxidation of a fuel at the anode and the reduction of an oxidant at the cathode. The most widely investigated FCs use hydrogen as fuel and oxygen (pure or within environmental air) as oxidants. Hydrogen FCs are already available in the market; in the future, they will play a relevant role in the decarbonization of transportation, residential usage, and industry [2]. However, hydrogen FCs require a complex balance of plant and are not suitable for powering, e.g., small portable devices [3]. Oppositely, fuels that are liquid at ambient temperature and pressure show the potential for portable power applications, as they allow operation at ambient pressure, with the advantage of a much easier storage [4].
Among the potential liquid fuels for FCs, alcohols have attracted most of the attention. This is due to the fact that they are thermodynamically unstable and, in principle, easy to oxidize with reduction potentials close to that of hydrogen [5, 6]. This basic fact guarantees that an FC fed with alcohol, known as the direct alcohol fuel cell (DAFC), can show open circuit potentials exceeding 1 V. The most investigated DAFCs use methanol (direct methanol fuel cells (DMFCs)) and ethanol (direct ethanol fuel cells (DEFCs)). The use of methanol and ethanol in FCs has several key advantages: (1) methanol and ethanol are easy to store and distribute; (2) they possess high volumetric energy density [7, 8]; (3) both can be obtained by fermentation or transformation of biomass and waste biomass with environmentally friendly processes that contribute to circular economy [9].
However, there are critical issues that prevent the commercial exploitation of DAFCs. Alcohols have much lower mass-weighted energy density than hydrogen [10, 11]. Being alcohols relatively more complex compared to hydrogen, their electrooxidation has knottier kinetics requiring higher activation energy than hydrogen oxidation. The kinetics also affects the oxidation products. Indeed, while methanol is usually fully oxidized to CO2 or carbonates depending on pH values, the cleavage of the C–C bond in ethanol is difficult and the most relevant oxidation pathways lead to the transfer of only 2 or 4 electrons instead of the theoretical maximum number of 12 electrons. This aspect limits the Faradaic efficiency of the device, significantly reducing the ability of DEFCs to exploit the energy content of the fuel. Possibly, the complete oxidation of ethanol to CO2 is the most relevant kinetic issue in DAFCs [12]. Many researchers have indeed worked for developing electrocatalysts able to cleave the C–C bond in ethanol, but despite of some interesting advances and successful examples [13, 14], the complete oxidation is still the less probable pathway.
A further important drawback of DAFCs is the permeability of membranes to the alcohols; this phenomenon is known as fuel crossover. Crossover affects much the performance of DAFCs for two reasons: (i) the loss of fuel and (ii) the decrease of the cell potential because of the mixed potentials at the cathode [15]. In the latter case, the alcohol is oxidized at the cathode, with its electrocatalyst not selective for the sole reduction reaction but active toward the oxidation of alcohols. So far, important research has been carried out on the anodic electrocatalysis, and in parallel, important effort has also been spent on the cathodic reaction to avoid mixed potentials. In addition, membranes play a crucial role as both an ion conductor and a physical separator to create a barrier for diminishing the negative effects derived from the alcohol crossover.
In DAFCs, two leading technologies have emerged based on: (i) proton exchange membranes (PEMs) [16] and (ii) anion exchange membranes (AEMs) [17]. DAFCs based on PEMs have been developed mainly for the availability of commercial membranes (e.g., Nafion) previously developed for PEMFCs and inherited entirely from that technology. Later, the increasing availability of efficient commercial AEMs (e.g., Tokuyama) has opened new venues in developing alkaline-based DAFC systems. While the two systems share the same aim of providing reliable and easy handling power sources, they have different features and related issues. Among them, it has to be mentioned that, during the oxidation of alcohols, alkaline systems provide product mixtures in the form of dissolved cations (e.g., carbonates for methanol oxidation and acetate for ethanol oxidation). This implies that alkaline DAFCs require stoichiometric amounts of alkali in the fuel. Indeed, according to the complete reaction scheme of the cell, hydroxides (OH−) are consumed to balance the charge that forms from the oxidation of the neutral molecule of an alcohol to a negatively charge ion [18, 19].
However, alcohol oxidation in acidic conditions has a more demanding kinetics than in alkali [20]. Moreover, the use of alkaline electrolytes increases the durability of the components of the fuel cells. In fact, it is easier to tailor alkaline fuel concentrations to minimize bipolar plate degradation [21], carbon support dissolution [22] or anodic catalyst leaching [23]. Alkaline FCs also have a crucial advantage related to the cathodic reaction. Indeed, in alkaline media, the gap in the oxygen reduction reaction (ORR) electrocatalytic activity between platinum group metal (PGM) and platinum group metal-free (PGM-free) electrocatalysts is significantly reduced compared to in acidic systems, enabling the use of PGM-free electrocatalysts at the cathode. The utilization of PGM-free cathodes led to significant advantages, as PGM-free electrocatalysts are not active toward the oxidation of the alcohols and avoid potential drops (mixed potentials) due to the alcohol crossover from the anode to the cathode [24,25,26].
A comparative analysis of alkaline and acidic systems for DAFCs is currently missing in the existing literature. We believe that such comparison is of paramount importance to highlight the peculiarity of the systems. The purpose of this review is to present for the first time such a comparative analysis, also covering the recent advances on materials, operation, and technology of DAFCs. Moreover, so far, no analysis has been proposed on the effect of the nature of the oxidation products in acid and alkaline environment. Remarkably, in acids, the oxidation products are generally neutral, and the system operates with aqueous solutions of the alcohols in the anode compartment. Oppositely, alkaline systems require high concentrations of OH− that is consumed stoichiometrically. Accordingly, the stoichiometric OH− consumption happens to guarantee the reaction charge balance. This aspect must be considered in the design of the fuel of alkaline systems and may severely affect the energetics of the reactions. For example, it has been demonstrated that the kinetics of ethanol electrooxidation in alkali is affected more by the OH− concentration than by the ethanol concentration [27]. The need for using OH− also affects the energy evaluation of the system as the production of alkali that happens most with the chloro-soda industry is highly energy intensive and may contribute importantly to the energy assessment of the technology. This aspect has already been demonstrated by the analysis of the case of electrochemical reforming of ethanol [28, 29] but has not yet been considered for DAFCs.
This review provides an initial brief description of the components of the DAFC membrane electrode assembly (MEA), the core of the fuel cell assembly. A description of the reactions occurring on the anode and the cathode of acid and alkaline DAFCs is elucidated. The thermodynamics and kinetics of alcohol oxidation reactions in alkaline and acid conditions are described, focusing the attention on the main differences between the two operating conditions. Then, the materials currently used for the membrane electrode assemblies (MEAs), named electrolytic membranes and electrocatalysts (anodes and cathodes), are introduced and described with an emphasis regarding their actual limitations. Membrane technologies used in acid and alkaline DAFCs are examined, introducing the most relevant and promising polymeric materials. Notably, the progress achieved by utilizing PGM-free electrocatalysts substituting PGM cathode electrocatalysts is deeply discussed, and its extraordinary tolerance towards alcohol oxidation is presented. Advancements in terms of operating condition optimization and transport phenomenon mitigation/control are deeply reviewed. The state-of-the-art electrochemical performance in terms of power density output is reported and discussed. Durability studies are also presented and considered. Ultimately, strategies adopted for overcoming problems are displayed, and future perspectives and outlooks are provided and suggested drawing a possible roadmap towards wide commercialization.
1.1 The Core of a DAFC: The Membrane Electrode Assembly
The MEA is the core of the fuel cell. This first section will briefly summarize its main constituents (a precise description of the fuel-cell components and assembly can be easily found in the already published literature and is out of the purpose of this review). Minor MEA constituent changes can be addressed between different FCs, depending on the fuels adopted for the different half-reactions [30]. In general, the MEA is constituted by three distinct elements: (1) an electrolytic membrane; (2) the electrocatalytic layers; (3) the diffusion layers (Fig. 1). The electrolytic polymeric membrane is positioned at the center of the MEA; it is made by a polymeric material which acts as: (a) a separator between the anodic and cathodic compartments of the fuel cell and (b) an ionic conductor for certain species (H+ for proton exchange membranes, OH− for anion exchange membranes) which can migrate between the two compartments, closing the circuit and permitting the external current flowing in the cell. The membrane is also the prime responsible for fuel crossover, a detrimental phenomenon occurring in DAFCs, in which the fuel crosses the membrane from the anodic compartment, occupying catalyst active sites otherwise used for the cathodic reactions. The anodic and cathodic electrocatalytic layers are placed on the two sides of the membrane and present themselves in form of thin compact layer composed by a mixture of a finely dispersed electrocatalyst powders and ionomers. Particularly, DAFC electrocatalysts can be divided into two main and general groups: (1) metallic nanoparticles (for both anodic and cathodic reactions) supported over a carbon backbone and this is the typical case of platinum based electrocatalysts (Pt/C); and (2) electroactive functional groups of the type M–N–C with M being a transition metal such as Mn, Fe, Co and Ni grafted onto the backbone of the support material (mainly for the cathodic reactions [31]) and this is the typical example of PGM-free electrocatalysts. The MEA is completed by the two diffusion layers (DLs), placed externally, and in contact with the two electrocatalytic layers. DLs have four main features that are the desired requisites: (i) porosity, (ii) electrical conductivity, (iii) chemical stability, and (iv) mechanical stability. These specific characteristics are required to: (a) permit the flow of fuel/byproducts to/from the electrocatalysts, (b) act as a current collector for the external leads of the FC, and (c) grant structural stability to the MEA, by sandwiching the electrocatalysts at the two sides of the membrane and avoiding electrocatalyst loss into the fuel. Lab-grade FCs usually operate with carbon cloth or carbon paper as DLs, while industrial cell stacks exploit more rigid and durable metallic sponges or meshes for this particular purpose [32,33,34,35].

Schematic diagram of a direct alcohol fuel cell
In this review, the state-of-the-art research on both membrane electrolytes (Sect. 4) and electrocatalytic materials (Sect. 5) is reported.
2 Alcohol Oxidation Reactions in Alkali and Acid: Thermodynamics and Kinetics
2.1 Theoretical Reactions and Potentials
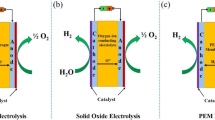
Figure 2a and b illustrates the schematic diagrams of DMFCs operating in acid and alkaline conditions, while Fig. 2c and d reports the schematic diagrams of DEFCs operating in acid and alkaline conditions. In acidic operations, the protons (more precisely the hydronium H3O+) move from the anode to the cathode while in alkaline operations, the hydroxides move from the cathode to the anode balancing the redox reactions. In both cases, the electrons move through the external circuit producing electricity. Figure 2 lists the theoretical half reactions and the overall reactions occurring in DMFCs and DEFCs operating in acid and alkaline environment, together with the relative potentials.

Schematic diagrams of a acid DMFC, b alkaline DMFC, c acidic DEFC and d alkaline DEFC, together with the main reactions taking place in the cell, and their potentials (expressed as V vs. the SHE for the half reactions). The reactions are for acidic DMFCs [36], alkaline DMFCs [37, 38], acidic DEFCs [36, 37, 39], alkaline DEFCs [38, 40, 41]. The molecules are not in scale
2.2 Thermodynamic Considerations: Theoretical Cell Potentials in Acid and Alkaline Conditions
According to Fig. 2, the theoretical cell potential (E0) for methanol that undergoes complete oxidation both in acid and alkali is practically the same at pH 0 and pH 14. The only difference is that, in acid, the oxidation leads directly to CO2, while in alkali at pH 14, the formation of carbonate leads to a reduction in valuable potential. The situation varies importantly when ethanol is the fuel. Indeed, partial oxidation may lead to significant changes in the cell potential. In DEFCs, if we assume a complete conversion to CO2, the theoretical cell potential in acid is 1.145 V versus the RHE. However, such a complete conversion is a minor pathway. The most likely products coming from the oxidation of ethanol are acetaldehyde and acetic acid. A cell that would produce solely acetaldehyde would generate a maximum potential of 1.032 V, slightly lower than that of the complete conversion to CO2. However, acetic acid is an important product in these conditions and a cell that would oxidize ethanol selectively to acetic acid would lead to a potential of 0.432 V with a more than two-fold reduction compared to the oxidation to CO2. In real systems, as the oxidation results in a distribution among 2, 4 and 12 electrons, the thermodynamic potential will be a combination of the previously mentioned values, resulting in a potential which is significantly lower compared to the theoretical value. In alkaline DEFCs, the complete oxidation of ethanol leads to the formation of carbonate. A cell capable of converting ethanol to carbonate has a theoretical potential of 1.171 V, slightly higher than the potential of acidic cells. However, the dominant pathway in alkali is the one that leads to the formation of acetate that still possesses an important theoretical potential of 1.121 V.
2.3 A Breakdown of Direct Alcohol Fuel Cell Potentials
The overall potential (E) that an FC could practically deliver is lower than its theoretical potential (E0). This is due to a series of efficiency loss contributions summarized in the term of overpotential (\(\eta\)). These contributions can be consequent to fundamental and technological factors. Electrolyte and fuel concentration, fuel crossover, reaction overpotentials and reaction pathways could be named among the fundamental factors that affect the cell potential. Fuel crossover on PGM cathodic electrocatalysts is one of them; this could impair ORR efficiency by the occupation of the cathodic catalyst active sites by the alcohol molecules. On the other hand, the main technological limitations could be directly ascribed to the architecture of the MEA. The sandwich structure that composes the cell, and the nature of the electrolytic membrane are in fact the main cause for the ohmic resistance (named as iR drop), and for the flux of chemicals to/from the electrode surfaces. All these factors are considered in the overall overpotential \(\eta\) as a linear combination, as stated in Eq. (1) [42]:
where \(\eta\)m takes into consideration the mixed potentials due to fuel crossover, \(\eta\)k is the activation overpotential dependent on the chemical nature of the electrocatalyst and on the distribution of its active sites, \(\eta\)r is the overpotential due to iR drop taking into consideration all the electrical losses (mainly due to technological factors), and \(\eta\)t is the overpotential due to fuel transport phenomenon (the depletion of the fuel concentration in the solution, hindering the reactive species in reaching the electrocatalyst). The different \(\eta\) terms have different weights, which are strictly dependent on the extracted current. At low currents, an increase in \(\eta\) is caused by the electrocatalyst activation losses, while at high currents the depletion of electroactive species from the double layer plays a major role and could induce a strong increase in overpotential. A constant contribution to the overpotential could be attributed to the mixed potential effect, while a linear increase in \(\eta\) with the current (i) can be finally expected by the iR drop component. The influence of all the parameters on the overall i–V curve of alkaline fuel cells as an example is summarized in Fig. 3, together with the single contributions.

Cell potential distribution (anode activation, cathode activation, iR drop, mass transport) for alkaline fuel cells
2.4 Alcohol Oxidation Kinetics in Acidic and Alkaline Conditions
Figure 2 reports the oxidation potentials for the complete and the partial oxidation of the alcohols considered and, in this case, methanol and ethanol. While these reactions well schematize the oxidation of a C1 alcohol as methanol, they did not represent a real screenshot of the reactions that are actually occurring in the fuel cells operating with alcohols with 2 or more carbon atoms (e.g., ethanol, glycerol, etc.). Unfortunately, as stated in Sect. 2.2, the oxidation is more often partial than complete, in other words, not all the electrons are harvested from the initial molecules and this, in turn, also reflects negatively on the actual thermodynamic potential of the systems. In the following sections, the expected different mechanisms for methanol oxidation reaction (MOR) and ethanol oxidation reaction (EOR) are reported.
2.4.1 Methanol
Methanol oxidation reaction (MOR) is still a challenging reaction despite the relative simplicity of the theoretical reaction pathway, which does not involve C–C bond cleavage. Methanol reaction which leads to complete oxidation to CO2, as reported in Fig. 2, proceeds according to Eq. (2).
Unfortunately, the oxidation mechanism is rather complex and, according to Leger [43], it involves eleven possible steps. Still today, the most effective electrocatalysts for the MOR are based on noble metals, especially Pt and its alloys (e.g., Pt–Ru). A short survey of the mechanism for the MOR in acidic conditions on Pt as the state of the art for DMFCs is presented. The first step of the mechanism is the adsorption of a methanol molecule on the Pt, according to Fig. 4 Reaction A. After the adsorption step, two main pathways (Reaction B1 or B2) can be taken by the molecule: the first step after adsorption is the formation of radical species with the release of 1 e− according to Fig. 4. Further monoelectronic oxidation (Reaction C1 or C2) occurs. Both the products of Reactions C1 and C2 can be oxidized to Pt-(CHO)ads through Reaction D1 or D2. The Pt-(CHO)ads is then oxidized to Pt-(CO)ads according to Reaction E (Fig. 4). This reaction is responsible for the poisoning of Pt-based catalysts in DMFCs as CO is so strongly adsorbed to Pt blocking the active sites, thus hampering further oxidation. In parallel with the formation of the formyl like species and CO adsorption, OH adsorption could occur as well at the Pt surface (Reaction F, Fig. 4). Adsorbed OH species may react with the adsorbed formyl to (i) directly produce CO2 (Reaction G) or (ii) form adsorbed COOH groups (Reaction H) which, according to Reaction I, are successively oxidized to CO2. To oxidize CO, larger overpotentials are required. Under such conditions, production of CO2 occurs directly via Reaction J or indirectly via Reaction K followed by Reaction I (Fig. 4).

Methanol electrooxidation reaction mechanism [40]
This discussion on the mechanism is limited to the acidic environment, as in these operating conditions, platinum exhibits the lowest overpotentials for the MOR, thus preventing the electrocatalyst poisoning, and prolonging the life of the electrocatalyst. Platinum is often alloyed to ruthenium for the MOR reaction, due to a synergistic effect between the two metals. Indeed, platinum is very active for the dissociative chemisorption of methanol, while the oxidation of the carbonaceous adsorbate to CO2 is favoured by the presence of the oxidized form of ruthenium [44]. At present, platinum-ruthenium alloys are the state-of-the-art electrocatalysts for the MOR.
2.4.2 Ethanol
Ethanol is receiving much attention for its exploitation as fuel in DEFCs, largely for its renewable nature, its well-established distribution infrastructure and lower toxicity as compared to methanol. Figure 5 reports the variety of oxidation products attainable in principle through electrocatalysis. The pathway which leads to complete oxidation to CO2 would give 12 e− but it is challenging to obtain because it implies the breakage of the C–C bond. It is then much commoner to obtain acetic acid and acetaldehyde (Reactions A, B and C in Fig. 6), delivering 4 e− and 2 e−, respectively [45].

Possible ethanol oxidation pathways. Readapted with permission from Ref. [1]. Copyright © 2013, Springer

As pointed out, complete oxidation is difficult to obtain. Next, a description of the complex mechanism leading to the formation of the above-mentioned products is reported. Aldehydes may react through the following pathway to render acetic acid (Reactions C and E in Fig. 6): CO2 is difficult to obtain; this pathway leads to the formation of methane from the adsorbed aldehyde (Reactions F and G). The adsorbed methyl may recombine with adsorbed hydrogen (produced by water adsorption, Reaction D1) to produce methane, freeing both catalytic metal sites (Reaction I). On the other side, CO may react with the hydroxyl adsorbed at the platinum surface to produce CO2 (Reaction H).
It is important to highlight the role that the adsorbed hydroxyl species play in the oxidation of ethanol. As for the methanol oxidation, the presence of adsorbed CO species at the platinum surface may hamper the electrocatalytic activity. Nevertheless, its occurrence is essential to producing a full oxidation to CO2. Indeed, it is the presence of adsorbed hydroxyl which allows CO to be oxidized to CO2. Coupling materials capable of increasing the rate of formation of the adsorbed hydroxyl at the electrocatalyst surface are indeed a key for increasing the effectiveness of the ethanol electrooxidation.
Lately, ethanol electrooxidation has been widely explored in alkaline media mainly because Pt can be effectively substituted by Pd indeed leading to even higher performance. Nevertheless, the C–C bond cleavage in alkaline environments has proved to not occur for ethanol at pH > 13 [46], acetate being the only oxidation product according to Eq. (3).
To obtain the full oxidation to acetate, the adsorption of hydroxyl is essential. Indeed, it has been demonstrated that palladium hydroxyl adsorption is the rate determining step [27], at least at low overpotentials. The addition of materials which may increase the hydroxyl adsorption rate on palladium has proved to be effective in enhancing ethanol electrooxidation. This is the case of Ceria, which is capable of improving the kinetics via spill-over of the primary oxide [47].
2.5 Fuel Composition: Acidic Versus Alkaline
Figure 2 summarizes the complete and half reactions occurring in acidic and alkaline systems. The reactions show that in acidic conditions, the products are always neutral (without charge), while in alkali, they are in form of anions. For methanol, the oxidation in alkali leads to the formation of carbonate with two negative charges; accordingly, two moles of OH− are required to oxidize one mole of methanol. Concerning ethanol, the partial oxidation to acetate requires an equimolar consumption of the alcohol and OH−. In passive systems, where the fuel is held in a tank directly in contact with the anodic electrocatalyst, the consumption of the fuel leads to the drop of potential with the time. In active systems, where the fuel is continuously renewed and flowing at the electrocatalyst, local variation in the thermodynamic conditions can occur in the flow field, with a more significant effect in alkaline systems compared to acidic ones. Remarkably, in the case of oxidation to carbonate, OH− depletes more rapidly than the fuel. Indeed, the oxidation of one mole of ethanol provides two moles of carbonates with the consumption of 4 OH− for each mole of ethanol. Accordingly, this affects the thermodynamics of the cell more than the molarity of alcohol itself. This fundamental aspect has been poorly highlighted in the literature; however, it is of crucial importance as it has important significance on the system engineering. The fuel must contain stoichiometric amounts of OH−, possibly exceeding the concentration of the alcohol. This consideration is due to the faster oxidation kinetics in the presence of the significant amount of OH− [48], especially in the low overpotential regions where, at least for ethanol, the oxidation kinetics is entirely dominated by the OH− concentration on the electrode surface [49].
2.6 Device Energy Density
As liquid fuels, alcohols show a relatively high energy density. Table 1 summarizes the energy density of ethanol and methanol compared to common fuels. The comparison shows that alcohols have a weight energy density roughly 30% lower than gasoline or diesel. There are important differences between methanol and other alcohols with two or more carbon atoms. Indeed, the energy density of methanol is lower. Oppositely, alcohols show an undoubtful advantage when compared to hydrogen having much larger volumetric energy density, especially at low pressure.
However, this is a point in favor of alcohols only in principle. Indeed, there are factors hampering the capability to convert the energy of the fuel into electric energy. In 2012, a study aiming to investigate the system efficiency was conducted on alkaline devices for ethanol oxidation in passive systems [50] showing a maximum efficiency of 12%. For DMFCs, efficiency up to 40% has been reported [51]. The main reason for this loss of efficiency is the higher complexity of the fuel molecule (ethanol > methanol > hydrogen), requiring more steps for the full oxidation.
The main reason why DEFCs have much lower efficiency compared to DMFCs is the difficulty in breaking down the C–C bond through electrochemical methods. For alcohols with two or more carbon atoms, the efficiency is seldom reported. As a result, it was calculated and found that, in alkaline systems, the energy efficiency topped the 12% even at a relatively low power generation [50]. This means that only the 12% of the energy contained in the fuel is transformed, a fact that overshadows the potential advantage of a high-density liquid fuel as compared, for example, to pressurized hydrogen. This is particularly the case for high power application, while it may leave open a potential exploitation for small power sources such as, for example, for portable or emergency applications.
3 Electrolytes
3.1 Ionic Conducting Membranes for DAFCs
Polymeric membranes play a crucial role within DAFCs working as physical separators between anode and cathode reagents/products and as ion conductors to allow protons flowing from the anode to the cathode (PEM) or anions flowing from the cathode to the anode (AEM). The desirable properties of a material to be chosen as a membrane for FC application are: (1) high ionic conductivity and zero electronic conductivity; (2) electrochemical and chemical stability at the operating temperatures (typically \(\geqslant\) 80 °C) in both oxidizing and reducing environments; (3) high mechanical durability; and (4) adequate physical barrier to oxidant and fuel crossover. Moreover, the choice of the electrolyte must be done considering its cost and readily availability as well as its environmental impact, preferring easy disposable, biodegradable and non-hazardous materials for operators and hand-users.
Proton conducting membranes tested in DAFCs can be classified in perfluorinated sulfonic acid membranes, non-perfluorinated membranes and their corresponding inorganic and organic composites or blends. In parallel, for alkaline DAFC technologies, anion conducting membranes are instead employed as electrolytes.
3.2 Nafion® and Other Perfluorinated Sulfonic Acid Membranes
Nafion® (DuPont) has been the most utilized polymer membranes in DAFCs. Nafion® consists of a polytetrafluoroethylene (PTFE) backbone and regularly spaced long perfluorovinyl ether pendant side chains terminated by sulfonic functional groups. The general chemical structure of Nafion® is illustrated in Fig. 7. The hydrophobic polytetrafluoroethylene backbone of Nafion® provides thermal and chemical stability, whereas the perfluorinated side chains terminating with hydrophilic sulfonic acid (–SO3H) act as proton-conductive groups. Protons go from the anode to the cathode of the MEA moving through the hydrophilic channels providing a relatively high ionic conductivity to the membrane (i.e., 90–120 mS cm−1 at 80 °C under a relative humidity ranging from 34% to 100%, see Refs. [52, 53]). However, Nafion® has several disadvantages when used in DAFCs, such as high production cost, low conductivity at low humidity or high temperatures, loss of mechanical stability at high temperatures, restricted operating temperatures and elevated alcohol permeability and Ru crossover when Pt-Ru bimetallic electrocatalysts are used for alcohol oxidation [54]. Alcohol permeability is one of the most challenging issues in DAFCs, since alcohol permeation at the cathode induces mixed potential effect with consequent decline of O2 reduction efficiency. The high alcohol permeability explains why Nafion® 115 and Nafion® 117 are usually preferred to Nafion® 112 widely employed in PEMFCs, since their high thickness reduces the alcohol crossover (see Table 2) [55, 56].

General chemical structure of Nafion®, where k = 6–7, m = 1, and n = 1
Nafion® membranes are susceptible to rapid dehydration at elevated temperatures, which results in the loss of conductivity and in some cases irreversible changes in membrane microstructure. Thus, one of the major challenges in the current research is to develop alternative membranes with the ability of working under elevated temperatures or low humidification of reactant gases.
Several companies have developed other fluorinated commercial membranes with structures similar to Nafion® but with different values of k, m and n, such as Hyflon® (Solvay Solexis), Flemion™ (Asahi Glass Engineering, Japan), Gore-Tex and Gore Select (Gore & Assoc, USA), 3P membranes (3P Energy, Germany) (see Table 2). The latter are perfluorinated sulfonic acid polymers showing a methanol permeability 20 times lower that Nafion® membranes allowing to operate with higher alcohol concentration resulting in turn in a higher power density [57]. However, the lack of other information in the literature does not allow to have a complete view about other key properties such as mechanical durability and lifetime of these membranes. Hyflon® (now commercialized with the name Aquivion from Solvay Specialty Polymers Italy) is a short-side-chain proton conducting perfluoropolymer membrane, characterized by excellent chemical stability and equivalent weight (i.e., grams of dry polymer per mole of ion exchange sites) of 850 g eq−1, lower than conventional Nafion® 117 (1 100 g eq−1) ionomer-based membrane. Its application in DMFCs is reported in Ref. [58] with a high-power density even if the details about long-term stability are not reported. The fluorinated membranes of other companies have been tested as polymer electrolytes for H2 fed FCs [57, 59,60,61], and their possible application in DAFCs requires efforts to modify the membranes to face the challenges typical of using alcohols as fuel.
Nafion-like membranes where –COOH weak acid groups replace –SO3H groups are also available, like Aciplex (Asahi Kasei, Japan) and membranes prepared with copolymers of tetrafluoroethylene and vinylene, such as the XUS membranes (Dow Chemical, USA). With its short side-chain the Dow membrane behaves very differently when compared to Nafion®. The specific conductance of the 800 and 850 EW membranes is 0.2 and 0.12 Ω−1 cm−1, respectively [62]. They are more permeable to methanol than Nafion® 117 but are 50 μm thinner. Experimental tests showed approximate values of current density due to methanol crossover of 4.0 × 10−10 and 2.7 × 10−10 A cm−2 for the XUS and Nafion® membranes, respectively [62]. However, to the best of our knowledge, there are no papers reporting DMFC performance data for the Dow membranes.
3.3 Composite Fluorinated Membranes
The creation of composite membranes is an effective approach to improve the performance of perfluorinated membranes. The strategy is to introduce inorganic compounds in the perfluorinated polymer with the aim to reduce methanol crossover, or acidic-basic polyaryl to reduce methanol crossover and increase conductivity.
The properties of hybrid organic–inorganic membranes are largely determined by the interactions in the mineral-organic interface [63]. Several inorganic fillers were proposed and tested to prepare Nafion® composite membranes and among them noble metals have deserved attention in relation to applications in DAFCs (mainly DMFCs). Noble metals such as Au, Pd, Pd and Ru are employed as nanoparticles or 1D nanostructures or as thin films to provide an effective barrier layer onto the electrolyte surface [54, 64,65,66,67,68,69,70,71,72,73,74]. The efficacy of this strategy depends on the noble metal load, distribution, and stability and in the case of thin films by the mechanical properties of the layers.
Another widely investigated filler is SiO2 employed to prepare Nafion® membranes as received or after functionalization [73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88], or as organic silica [75,76,77,78,79,80,81]. The addition of these fillers to Nafion® can improve thermal stability, proton conductivity and, notably, resistance to methanol crossover [54, 61, 65,66,67,68,69,70,71, 75,76,77,78,79, 82,83,84,85,86,87,88,89,90,91,92,93,94,95,96]. Zeolites [82,83,84,85,86,87,88,89,90,91], montmorillonite [92,93,94,95,96,97,98,99,100,101,102], silicate [103], aluminophosphate [103], aluminosilicate [104], calcium phosphate [105], zirconium phosphate [106,107,108,109,110,111,112], and zirconium sulphophenyl phosphate [113] were also tested as inorganic fillers able to minimize methanol crossover. Some Nafion® binary inorganic composites have been studied in relation to DMFCs, such as silicotungstic acid-SiO2 [114], phosphotungstic acid-SiO2 [114, 115], phosphotungstic acid-mesoporous SiO2 [116], and Pd-SiO2 [117].
Modification of Nafion® through the addition of silica is a common strategy for enhancing membrane performance in DMFCs. Nafion®-silica membranes have been prepared using silica powder [118,119,120], diphenylsilicate (DPS) [78], sol–gel reaction with tetraethylorthosilicate (TEOS) [121], SiO2 and phosphotungstic acid (PWA) [114]. Nafion®-silica membranes show good performance at T > 100 °C due to low levels of dehydration allowing to get peak power density up to 240 mW cm−2 for an oxygen-fed cell with a measured methanol crossover rate of 4 × 10−6 mol min−1 cm−2. Mesoporous SiO2 layers were exploited as well to modify the surface of Nafion® 117 membrane via a simple dip-coating technique [122] reaching a proton conductivity of 68.5 mS cm−1 at 80 °C with a methanol diffusion coefficient of 6.95 × 10−8 cm2 s−1, significantly lower with respect to the diffusion coefficient in Nafion® 117 estimated in the same condition. The modification of Nafion® membranes through the addition of molybdophosphoric acid (MoPh-a) has been shown to increase the proton conductivity 2.0–2.5 times, but with slightly increased methanol crossover [118].
Inorganic fillers such as zirconium phosphate (ZrP), a layered acidic inorganic cation-exchange material with the formula Zr(HPO4)2·2H2O, has been explored to improve the performance of Nafion®. Indeed, ZrP is known for its great thermal and chemical stability, as well as its high ion conductivity and mechanical strength, and can be integrated into Nafion®-based membranes [123]. Nafion®-zirconium membranes can be prepared by starting with an extruded film such as Nafion® 115, that can then be impregnated with zirconium phosphate (ZrP) via an exchange reaction involving zirconium ions followed by precipitation of zirconium phosphate by immersion of the membrane in H3PO4 solution. The result is an insoluble ZrP entrapped in the pores of the Nafion® membrane. Nafion® zirconium membrane is stable at T = 150 °C with dry oxidant. The membrane resistance was measured to be 0.08 Ω cm2 and maximum power densities of 380 and 260 mW cm−2 for a DMFC with this membrane were achieved with oxygen and air feeds, respectively [107]. The ZrP additive can enhance water retention, raise the maximum working temperature, and increase the dry weight and thickness of the membrane by 23% and 30%, respectively [107, 124]. More recently, other attempts of ZrP composite membrane fabrication led to comparable performance in terms of power density [125,126,127].
Nanocomposite membranes have been synthesized by in situ polymerisation of furfuryl alcohol (PFA) within commercial Nafion® membranes to reduce hydrophillicity of the latter. Furfuryl alcohol is miscible in water and alcohol mixtures (it penetrates the hydrophilic channels of Nafion®) and becomes hydrophobic following polymerization via acid catalysis. The Nafion®-8 wt% (wt% means the weight percentage) PFA membrane has a methanol crossover of 1.72 × 10−6 mol min−1 cm−1 and a proton conductivity of 70.4 mS cm−1 at room temperature. The corresponding properties of Nafion® 115 are 4.66 × 10−6 mol min−1 cm−1 and 95.3 mS cm−1 [128], thus the lower conductivity of Nafion®-8 wt% PFA is compensated by the significantly lower methanol permeability.
Composite membranes have also been prepared by using conductive polymers (i.e., polyaniline and polypyrrole), and introduced into Nafion® to reduce methanol permeability [128,129,130,131,132]. The membranes obtained by the electropolymerization of aniline at Nafion®-coated Pt electrode has lower ionic conductivity and lower methanol permeability compared with Nafion® membranes [130]. Sungpet developed an impure Nafion® membrane modified with polypyrrole [129]. The presence of polypyrrole in Nafion® membrane resulted in the decrease of the percentage of sorption capacity of methanol and the decrease of the methanol flux to the one third of that of Nafion®. Smit et al. developed a modified Nafion® membrane with in-situ polymerized polypyrrole with reduced methanol permeability [131]. The electrochemical modification of Nafion® membranes with polyaniline results in the reduction of one order of magnitude in methanol permeability at the cost of a slight decrease of their proton conductivity [132].
Among the composite fluorinated membranes, it is important to mention Pall IonClad® membranes, produced by Pall Gelman Sciences Inc., which are based on tetra-fluoroethylene/perflouropropylene. IonClad® R-1010 (36 μm thick) and IonClad® R-4010 (63 μm thick) have 2.5–3.0 times lower methanol crossover than Nafion® 117 (180 μm thick) at 60 °C and a comparable conductivity at 20 °C [133].
3.4 Non-fluorinated Membranes
Several non-fluorinated alternative polymers have been proposed for DAFCs, mainly based on sulfonated ionomers with an aromatic or aliphatic hydrocarbon skeleton [134]. The main advantage compared to Nafion® relies in avoiding the utilization of fluorinated compounds that are toxic especially at their end of life. Among them sulfonated polyether ether ketone (sPEEK) has been widely investigated due to their good mechanical properties, thermal and chemical stabilities [135]. Moreover, sPEEK is relatively cost-effective [136] and its ionic conductivity could be easily controlled by the degree of sulfonation. Narrow and more branched pores of sPEEK showed less methanol permeability in DMFCs in comparison with Nafion® [137]. However, excessive swelling of sPEEK due to a high degree of sulfonation may hamper its long-term use in DMFCs [138]. Many attempts have also been made to prepare polyblends. Wu et al. prepared sPEEK-poly (vinylpyrrolidine) blend membranes that could decrease the methanol crossover in DMFCs without affecting the ionic conductivity [139]. To reduce the methanol crossover and increase the ionic conductivity in DMFCs inorganic oxides (SiO2, ZrO2, TiO2, etc.), phosphates and heteropolyacids were incorporated into sPEEK [140,141,142]. The modification of sPEEK with methane sulfonic acid and zeolite 4A (sPEEK-MSA-zeolite 4A) results in an improvement of ionic conductivity and decrease in methanol crossover [143]. The addition of inert (no charge/hydrophobic) polyphosphazene to sPEEK was also studied to assess the effect on methanol crossover in DMFCs [144]. Other acid–base non-fluorinated polymers tested as proton conducting membranes in DMFCs are sulfonated polyphosphazene membranes [145] and sulfonated poly(ethylene-alt-tetrafluoroethylene) membranes [146].
Other widely studied non-fluorinated membranes are those based on polybenzimidazole (PBI), a non-ionic polymer that becomes a proton conductor when doped with a strong acid (i.e., the sulphuric or phosphoric acid [147]). Indeed, PBI is a basic polymer (pKa = 5.5) and doping with acid forms a single-phase polymer electrolyte that has a good oxidative and thermal stability, and mechanical flexibility at elevated temperatures T < 200 °C [148]. At T > 100 °C, these membranes possess high proton conductivity, low electroosmotic drag (ca. 0 compared to 0.6 for Nafion®), low methanol crossover (an 80-μm-thick PBI membrane has a methanol crossover of 1/10 with respect to that of a 210-μm-thick Nafion® membrane) [148]. The major disadvantage is the leaching of the acid used for the doping once exposed to methanol solutions at high temperatures. Better performance can be achieved by using a silica impregnated phosphoric acid doped polybenzimidazole (PA/PBI/SiO2) composite membrane [149]. The composite membrane shows a proton conductivity of 29–41 mS cm−1 at temperatures between 200 and 250 °C allowing DMFCs delivering a peak power density of 136 and 237 mW cm−2 at 260 °C using the Pt/C and PtRu/C as the anode electrocatalysts, respectively.
Sulfonated polyimides (sPIs) are another type of ionomers extremely suitable as polymer electrolyte membranes (PEMs) for fuel cell applications, except for their poor water stability. Crosslinking is a method that is commonly used to improve the weak hydrolytic stability of sPI membranes allowing to get performance in DMFCs comparable to that of Nafion® 212 [150].
Polyvinyl alcohol (PVA) based membranes are one such class which has receiving increasing attention in DMFC application [151]. Because PVA is not a proton-conducting polymer, it should be sulfonated by using different sulfonic-carboxylic acids forming crosslinked polymers. The research efforts on this kind of membranes as well as their performance in terms of methanol crossover and proton conductivity are summarized in [152, 153].
Non-fluorinated acid membranes can be also prepared in blend with polyvinylidene fluoride (PVDF), whose mechanical strength, thermal stability and good chemical resistance make it suitable to improve the properties of sulfonated polymers. Blend of PVDF with sulfonated polyimide [154], sulfonated polyether sulfone [155], sulfonated polyether ether ketone [156], and sulfonated poly [bis(benzimidazobenzisoquinolinone)] [157] has been used as polymer electrolyte for DMFCs. More recently, copolymers of poly(methyl methacrylate)-co-poly(2-acrylamido-2-methyl-1-propane sulfonic acid) (PMMA-co-PAMPS) have been blended with polyvinylidene fluoride (PVDF) and sulfonated PVDF (S-PVDF) to prepare proton conducting membranes [158], showing low methanol permeability (22 × 10−7 cm2 s−1).
3.5 Anion Exchange Membranes for Alkaline DAFCs
Alkaline DAFCs are very promising due to the several advantages with respect to the acidic counterparts. Indeed, the kinetics of both anodic oxidation of alcohols and cathodic oxygen reduction in alkaline media is more favourable than in acidic solution. Moreover, in alkaline media the cathodic catalysts are more methanol tolerant and the methanol crossover due to electro-osmotic drag would be reduced in OH− conducting membranes [22, 159]. Anion exchange membranes (AEMs), which act as both the ionic conductor and electronic insulator, need to have high hydroxide conductivity, high electrochemical and mechanical stability [160,161,162].
Some commercially available AEMs have been used in DAFCs. MORGANE®-ADP membranes from Solvay, ordinarily used for salt electrodialysis, were used as the solid polymer electrolytes for DMFCs [163,164,165,166]. They are cross-linked fluorinated polymers, whose exchange group is quaternary ammonium. They have thickness ranging between 150 and 160 μm when fully humidified with a resistance of 0.5 Ω cm2 in 1 M (1 M = 1 mol L−1) NaOH [164]. Encouraging performance were obtained, with the highest power density of 20 mW cm−2. They show to be less permeable to methanol compared to Nafion®, but they are not very stable in strong alkaline media.
Tokuyama Co. Japan is studying few ammonium-type AEM membranes and particularly, A201 and A901 were used for alkaline DEFCs [167,168,169], while AHA has been tested in DAFCs by using other alcohols [170].
Ongoing research on AEMs is mainly focused on polyvinylidene fluoride (PVDF, –[CH2CF2]n–) and poly(tetrafluoroethene-co-hexafluoro propylene) (FEP, –[CF2CF2]n[CF(CF3)CF2]m–) films grafted with vinylbenzyl chloride using radiation-grafting [171, 172]. The FEP membranes showed good properties suitable for the fuel cell application, but their cost is relatively high. Cationic polymers with high ion exchange capacity (IEC), such as quaternized cationic polymers, are the preferred materials for the preparation of AEMs with high hydroxide conductivity [173, 174].
Many kinds of AEMs based on quaternized polymers containing a quaternary ammonium group have been developed and tested in DAFCs, such as polysiloxane containing quaternary ammonium groups [175], aminated poly(oxyethylene) methacrylates [176], quaternized polyethersulfone cardo [177], quaternized poly(phthalazinon ether sulfone ketone) [178], and quaternized polyvinyl alcohol [179]. The best performance obtained with the latter in DMFCs and DEFCs with utilizing QPVA-based membranes allowed to get peak power densities of 272 and 144 mW cm−2, respectively.
An alternative approach to the production of AEMs is doping or reacting polymer films with KOH. Polybenzimidazole membrane was doped in alkaline solution and showed at room temperature higher conductivity than acid doped PBI [180]. MEAs prepared by using such membranes yielded power densities of ca. 30 mW cm−2 for methanol [181] and 60–70 mW cm−2 for ethanol [182]. Even higher peak power densities (125 mW cm−2) are reported for alkali-doped PBI in a DEFC using Pt-free catalysts [183]. Recently, in-situ crosslinked, side chain polybenzimidazole-based anion exchange membranes for alkaline direct methanol fuel cells allowed to deliver a maximum power density of 152.6 mW cm−2 using 5 M KOH and 3 M methanol fuels at 80 °C [184].
Polyvinyl alcohol composite membranes for use in alkaline DAFCs have also been investigated [185,186,187,188]. Polyvinyl alcohol (PVA) is a cheap synthetic polymer with selective permeability of water molecules over methanol molecules [189, 190]. PVA based blend and nanocomposites were prepared for alkaline DMFC operations [191,192,193]. Good performance has been obtained by using silica/PVA-pyridine hybrid anion exchange membranes thanks to the good thermal stability, mechanical properties, OH− conductivity up to 9.6 × 10−2 S cm−1 at 80 °C [194], and with PVA-based block copolymer successfully employed in DMFCs with a maximum peak power density of 99.6 mW cm−2 [195].
4 Electrocatalysts
4.1 Anode Electrocatalysts for Acid DAFCs
Pt is the most promising material among the pure metals for application in DAFC anodes because it exhibits the highest activity for the dissociative adsorption of alcohol (methanol or ethanol). Yet, pure Pt electrocatalysts are easily poisoned by CO, which is an intermediate of the methanol/ethanol oxidation reaction [196,197,198,199]. The electrocatalytic activity of Pt depends on structure, morphology, particle size and surface oxidation states [200,201,202,203]. The addition of Ru to Pt has proven effective in overcoming the poisoning of active sites by CO, due to OH groups generated on Ru sites (bifunctional mechanism) [204,205,206,207,208]. At present, three types of PtRu electrocatalysts can be found in the literature: PtRu alloy [209, 210], Ru@Pt core–shell structure [211, 212] and Pt modified with Ru atoms or clusters [213, 214]. The highest activity for methanol oxidation for a Pt–Ru electrocatalyst can be observed when Ru is in a solid solution with Pt [215]. An optimal Ru content of 50 at% (at% means the atomic percentage) was found for the MOR at temperatures from 90 to 130 °C [216]. At room temperature, methanol oxidation is faster on pure Pt–Ru alloys having low Ru content (10 at%); whereas, at intermediate temperatures (60 °C) the process is faster on alloys with 33 at% Ru content [217].
Pt-Ru cost is one of the main obstacles to the scaled application of DMFCs [218]. This issue can be addressed by modifying Pt electrocatalysts with cost-effective metals or metal oxides able to form surface-oxygenated species at lower potentials. Until now, almost all the transition metals (such as Ru, Rh, Pd, Os, Ir, Sn, Ni, Cu and Au.) have been alloyed with Pt and investigated for MOR [219,220,221,222,223,224,225]; among these, Ru has been found to be the most active. To reduce the amount of noble metals and increase the catalyst utilization, the approach of dispersing active nanoparticles onto suitable conductive supports, such as carbonaceous materials, has been used [226,227,228] (several researchers have also pursued the synthesis of multiple-component electrocatalysts such as PtRuM [229,230,231,232,233,234,235,236,237,238,239,240] and PtMOx (M = Ti, V, Mn, W, Ni, Mo, etc.) [241,242,243,244]). Furthermore, integrating Pt or PtRu with CeO2 [245, 246], TiO2 [247], IrO2 [248, 249], Ti0.1W0.9C [250], CoP [251], Ni2P [252] and heteroatom doped carbon [253,254,255] could also enhance the MOR activity. Increasing interest, as demonstrated by a huge number of publications in the last few years [256, 257], has been devoted to 1D nanoarchitectures, such as nanorods, nanowires, and nanotubes, able to provide high resistance to corrosion, large catalytic preferential facets or crystalline defects, which would deliver sufficient reaction sites with enhanced activity, open pores and interconnected channels, which would accelerate the mass transport of reactants (methanol) and products (carbon dioxide). Moreover, the advantage of these structures is related not only to mass transport, but also to improved conductivity. Recent results reported in the literature in the last decade concerning the electrocatalytic activity for MOR of new electrocatalyst formulations are shown in Table 3.
The ethanol oxidation reaction (EOR) is even more complicated and sluggish in kinetics than the MOR because it involves not only the ethanol dehydrogenation but also the C–C bond cleavage [258]. Accordingly, the efficiency of a DEFC is significantly limited by the formation of partial oxidation products (such as acetaldehyde and acetic acid) and strongly adsorbed intermediates [259]. The most investigated electrocatalyst, also in the case of EOR, is the binary Pt-Ru system, but other oxophillic metals (e.g., Sn or Rh), in combination with Pt, have been largely employed [13, 221, 260,261,262]. The most significant enhancement in DEFC performance has been observed in the case of Pt–Sn electrocatalysts [263], with the 3:1 atomic ratio being the most active for EOR [264]. As reported by Antolini and Gonzalez [265], the “state” of Sn metal phase (relative amount of Pt–Sn alloy and SnO2) affects significantly the DEFC performance in terms of electrochemical activity (and maximum power density). The most significant recent advances in electrocatalytic activity for EOR are reported in Table 4. High mass activity is essential for reducing the usage of noble metals in DMFCs and DEFCs. Specific activity is obtained from MOR/EOR current normalized by the electrochemically active surface area (ECSA), indicating the intrinsic activity of electrocatalysts.
As reported in Tables 3 and 4, many electrocatalytic systems show high activity for MOR/EOR; yet much work remains to be done to accelerate DAFC commercial applications. It must be considered that three-electrode cell results give information about the electrocatalytic activity for a specific reaction, excluding many other factors (such as conductivity of the catalytic layer, three-phase boundary in the presence of solid electrolyte, CO2 removal from the catalytic layer, methanol crossover and water-flooding in the cathode) involved in real fuel cell environment. Therefore, to have a complete analysis of the suitability of an electrocatalyst, it is mandatory to test the performance in a real fuel cell.
4.2 Anode Electrocatalysts for Alkaline DMFCs
Pt and Pd are also the prime electrocatalyst selection for high pH electrolytes, especially when used jointly with other less noble metals or oxides for both methanol and ethanol electrooxidation.
Methanol oxidation requires the use of Pt based materials in acidic DAFCs. However, as alkaline conditions are less severe in terms of material corrosion, other materials can effectively be used. A recent review [302] has highlighted the materials that have been currently exploited for the oxidation of methanol in alkaline environment. Pt based materials are as effective in alkali as in acids. Remarkably, the addition of Ru to Pt does not improve the kinetics of MOR in alkaline media [303]. Bimetallic catalysts containing gold and silver have been explored with significant effects on the activity. Remarkably, in [303], it was shown that an Au3Pt catalyst has the same specific activity as that of a full Pt catalyst. At first, gold might not be considered as suitable material since it is also a precious metal. However, the replacement of at least a portion of platinum with gold has a significant advantage. Indeed, gold, despite its cost which is currently higher compared to Pt, does not belong to the family of the critical raw materials (CRMs), while platinum is one of the most critical resources. This aspect has been highlighted by the recent reports on critical raw materials released by the European Commission [304]. To reduce the Pt loading, materials other than Ag and Au have been considered. So far, nickel appears to be one of the most promising. In [305], it was shown that low platinum alloys, PtNi3 and PtNi7, are extremely promising for MOR in alkaline media. PtIn alloys have also been explored. Santos et al. have tested MOR in catalysts with up to 50% atomic indium. They found that the best atomic ratio was 70:30 (Pt:In), attributing the improvement in the activity to the electronic effect and the occurrence of oxy-hydroxy interactions [306]. Palladium based materials are also effective for methanol oxidation in alkaline media. In [307], it was demonstrated that nanoparticles with a Pd shell can effectively oxidize methanol. Pd alloys in alkaline media were also considered in other studies with the purpose to determine selectivity and energy performance. In [308], the oxidation of methanol in alkaline electrolytes was investigated by electrochemical methods and high pressure liquid chromatography (HPLC) determining that the oxidation can be incomplete and leading to the formation of formaldehyde and formate. However, while Pd is less effective than Pt in the oxidation of methanol in alkali, the catalytic activity is improved with bimetallic Pd alloys. As pointed out in [309], the use of bimetallic Pd catalysts may significantly change this basic fact making Pd based nanoalloys competitive with Pt catalysts (e.g., Ni, Ag, Au, Pt, Sn) [310,311,312,313,314,315]. The use of bimetallic alloys might enable CO oxidation and removal from the surface of the electrocatalyst [309, 316], a key step in the complete electrooxidation of methanol. While more abundant than Pt, Pd is also in the list of critical raw materials and its use should be limited and whenever possible avoided [304]. To overcome the limitations related to the use of precious and rare resources, the use of electrocatalysts free of Pt and Pd has also been proposed. Nickel and nickel alloys and copper-based materials appear to be promising. In [317], Mansor et al. reviewed the state of the art of such materials that manifest significant current density even if they are still far from competing with the stability and the performance of the electrocatalysts based on Pt and Pd. Recent results reported in the literature in the last decade concerning the electrocatalytic activity for MOR of new electrocatalyst formulations are shown in Table 5.
4.3 Anode Electrocatalysts for Alkaline DEFCs
Much effort has been devoted to the development of anode electrocatalysts for ethanol in alkaline media. The subject has been extensively reviewed recently by Monyoncho et al. in [323] reporting an extensive table of the major catalysts along with the performance. As introduced in Sect. 3.4.2, ethanol electrooxidation is favored in alkaline media for both Pt and Pd based electrocatalysts [323] because of the increased presence of OHads on the surface of the electrode. The adsorbed hydroxide species might also promote the C–C bond cleavage (enhancing the Faradaic efficiency of the cell) [324, 325], aiding the transition from poisonous COads into CO2. However, at high pH values the major oxidation products especially on palladium catalysts remain acetate. In recent years, a wide research effort in the field of EOR electrocatalysis has mainly shifted towards alkaline electrolytes, for their better oxidation kinetics in respect to acidic systems.
Platinum, when used as a single metal electrocatalyst, works best for the complete oxidation of ethanol. This is more pronouced in alkaline electrolytes in respect to acidic ones [326]. It has been found that residual oxides, like the ones that can be found after the reduction of a Pt oxide film, increase the activity towards EOR by a factor of 30 [327]. However, it has been shown that Pt suffers from CO poisoning as well in alkaline electrolytes, limiting its stability and durability.
Studies performed on ordered monocrystalline surfaces showed a strong dependency of EOR performance and durability on the surface crystal terminations. It has been observed that surfaces with low coordination sites (e.g., high index facets) boost the oxidation current, and are less prone to deactivation [328] (e.g., on Pt(110) only adsorbed CO was observed whereas adsorbed CHx was only found on Pt(111) terrace sites). To overcome the problem of Pt deactivation, platinum was coupled with other elements or compounds, exploiting the bimetallic synergies. Pt was coupled with, e.g., W and Sn [329,330,331,332,333]. While less effective than in acids, the addition of ruthenium has also been considered [181, 329, 334].
Palladium was also deeply studied in the past decade as an alternative to platinum (at least until its cost per unit mass was lower in respect to Pt). Pd shows both advantages and disadvantages in respect to platinum. It is less prone to CO poisoning especially at pH values higher than 13, where the C–C bond cleavage is difficult, and the major oxidation product is acetate [335]. Also, the only product of ethanol through incomplete oxidation on palladium is acetate. This selectivity decreases the fuel efficiency of FCs, limiting Pd usage as standalone anodic catalysts. On the contrary, it has been seen that the conjunction of Pd with less noble, oxyphilic metals could favor the complete oxidation of ethanol. Carbon supported Pd materials have been largely investigated, delivering excellent energy performance in DEFCs [329, 336,337,338,339,340,341,342]. Palladium has also been applied to supports other than C. In [7], it was shown that titania nanotube arrays grown onto the surface of titanium non-woven webs can deliver excellent performance, with current densities higher than 1 A cm−2 and power densities exceeding 300 mW cm−2.
Binary and ternary Pd alloys have also been largely explored with beneficial effects to the electrocatalysis. Bimetallic alloys with Au, Sn, Ir, Ni, Ru and Cu were explored showing an improvement in current density and onset potential values compared to Pd alone [167, 186, 187, 336,337,338, 340, 343,344,345,346,347,348,349,350,351]. Ternary Pd alloys have also been considered. Notable examples are Pt–Ni–Sn, Pt–Ru–W, Pd–Ni–Zn and Pt–Ru–Mo [168, 329, 338]. Excellent activity enhancement has also been achieved by exploiting the coupling of Pd with promoting oxides (e.g., CeO2, NiO, SnO2). In [50], it was shown that the addition of CeO2 to a Pd/C catalyst produce a 100% increase in the cell efficiency at an even Pd loading.
An interesting approach to the improvement of the electrocatalytic activity of Pd based electrocatalysts for alkaline ethanol oxidation was proposed by Sun. In 2007, Tian et al. showed that THH palladium nanoparticles with (730) terminations show outstanding ethanol oxidation activity in alkali [352]. This investigation suggests that the engineering of the Pd particle crystal termination is an effective way to enhancing ethanol electrooxidation.
The major issue with Pd electrocatalysts seems to be the stability of the materials [19]. In the latest years, it has been shown that the oxidation of the palladium surface can compete with the ethanol oxidation reactions even in the conditions that the materials experience in fuel cells [353, 354]. Such oxidation can result in the partial dissolution of the nanoparticles that may lead to significant decrease of the surface area [355]. Such dissolution can be prevented by avoiding the exposure to excessive anodic stress, e.g., by avoiding the DEFC potential to drop below values that produce anode potentials larger than 0.6 V versus the RHE [19]. Pd oxidation can even be mitigated by the addition of small concentration of NaBH4 to the fuel as showed in [18].
Remarkably, a new approach to the electrooxidation of ethanol based on the use of supported organometallic complex has been proposed. In [47], an anode based on a Rh(I) diolefin amine complex was prepared and tested for the electrooxidation of ethanol both in half cell and in a complete fuel cell. The anode delivered a specific current density up to 10 000 A gRh−1. This high value was the result of using the organometallic complex spreading over the surface of a Ketjen Black carbon support. The fuel cells obtained with such an approach to the fabrication of the anode were defined as organo metallic fuel cells (OMFCs) [47]. Recent results reported in the literature in the last decade concerning the electrocatalytic activity for EOR and glycerol oxidation reaction (GOR) of new electrocatalyst formulations are shown in Table 6.
4.4 Cathode Electrocatalysts
Generally, the electrocatalysts of choice for the cathode of acidic DAFCs are restricted to two categories: Pt-based electrocatalysts (mainly Pt/C) and M–N–C PGM-free electrocatalysts (where M is a transition metal, mainly Fe or Co) [31]. Pt-based electrocatalysts have the advantage of showing higher performance, but they suffer from poor alcohol tolerance. On the other hand, the use of PGM-free electrocatalysts is limited to the M–N–C type, since transition metal oxides (e.g., Mn, Ni, and Co oxides) are generally unstable in acidic media and undergo corrosion [31]. M–N–C electrocatalysts show poor ORR performance due to their lower active-site density and turnover frequency. However, this drawback is compensated by their excellent alcohol tolerance, which makes them the optimal choice to mitigate the detrimental effect of alcohol crossover observed with Pt-based electrocatalysts.
For the cathode alkaline DAFCs, the electrocatalyst selection should fall on PGM-free materials rather than on PGM ones for three main reasons: (i) the performance gap between PGM-free and PGM electrocatalysts in alkaline media is generally much lower than in acidic media; (ii) PGM-free electrocatalysts show excellent alcohol tolerance; and (iii) PGM-free electrocatalysts show good stability in alkaline media. For example, unlike in acidic media, various transition metal oxides highly active towards ORR like Mn, Ni, Co, and Fe oxides (and their combinations) do not undergo corrosion and can be used at the cathode of alkaline DAFCs. However, the oxides are not very efficient towards the reduction of oxygen following the 4-electron pathway. In the end, PGM-free electrocatalysts show lower cost in respect to PGM materials, finally favoring the utilization of the former ones in respect to the latter ones at the cathode of alkaline DAFCs.
Transition metal oxides are generally easily synthesizable and cheap. However, compared to M–N–C materials, they show lower specific surface area, and they may suffer from poorer electronic conductivity. In the following sections, the electrocatalysts used for the cathode of DAFCs in acid and alkaline configurations are presented.
4.5 Cathodes for Acid DAFCs
In acid DAFCs, Pt-based electrocatalysts for ORR show good performance but unfortunately, they are also very active towards alcohol oxidation reactions. Thus, Pt-based electrocatalysts at the cathode are not effective considering the problem of the crossover of alcohol from the anode to cathode, which dramatically decreases the cell open circuit voltage. The formation of a mixed potential (oxygen reduction and alcohol oxidation occurring simultaneously on the active sites of the cathodic electrocatalyst), causes a significant performance loss when Pt/C electrocatalysts are used at the cathode of a DAFC. Figure 8 clearly shows the impact of alcohols on ORR in acid environment for a commercial Pt/C electrocatalyst and a Fe–N–C electrocatalyst [25, 31, 379,380,381,382,383,384,385,386,387]. The ORR activity of the Fe–N–C electrocatalyst is not affected by the presence of alcohols, demonstrating the high tolerance of PGM-free electrocatalysts towards the presence of alcohols at the cathode of DAFCs. It has been demonstrated that the presence of just 2 mM alcohols impacts significantly on Pt/C, reducing the ORR activity. When the concentration of alcohol reaches 200 mM, clearly the Pt/C catalyzes their oxidation, as visible by the decrease of the ORR current density towards the MOR and EOR. Thus, the development of alcohol tolerant ORR electrocatalysts is fundamental for the commercialization of DAFCs.

Effect of the presence of alcohols (methanol and ethanol) on ORR activity for a commercial Pt/C catalyst and a PGM-free Fe–N–C-catalyst, obtained in a half-cell configuration, 0.5 M H2SO4, 1 600 r min−1 (catalyst loadings: 50 mg cm−2 for Pt/C; 600 mg cm−2 for Fe–N–C). Adapted with permission from Ref. [25]. Copyright © 2017, Elsevier Ltd.
4.6 Cathodes for Alkaline DAFCs
The performance of alkaline DAFCs is usually superior compared to their acid counterparts, because of the faster kinetics of both oxidation and reduction reactions in alkaline environment. Moreover, in alkaline DAFCs, the alcohol crossover effect is mitigated by the OH− dragging alcohol molecules back to the anode side [388]. Nevertheless, selective and alcohol-tolerant ORR electrocatalysts are required to avoid as much as possible the cathode potential drop lowering the cell voltage [31]. PGM-free electrocatalysts are the suitable materials to be used at the cathode side. Generally, electrocatalysts in alkaline environment show a high stability compared to acid conditions [303, 388]. Table 8 summarizes the activity reached in recent years in alkaline DAFCs, fed either with methanol or ethanol.
5 Fuel Cell Performances
5.1 Effect of Membrane Electrode Assembly Configuration on Power Output
As no standard conditions have been selected by the scientific community and industries, MEA fabrication conditions are very much variable among the reported studies and often difficult to compare fairly. Importantly, the operating conditions significantly affect the power output and durability of DAFCs, independently from their operations in acidic or alkaline environment. In DAFCs, it is difficult to compare different power outputs, due to the different operating conditions. The MEAs fabrication procedure can also largely affect the power output. The MEA is formed by an anode electrode, a cathode electrode, and an electrolyte membrane. In turn, the electrodes are composed of a mixture of electrocatalyst, and ionomer applied over a DL. The variation of each component is crucial for an optimal power output. Considering the anode, as mentioned above, the electrocatalysts used are generally Pt/Ru supported over a carbonaceous matrix. It has been shown that anodic kinetics is quite poor and so an increase in anode electrocatalyst loading is important in improving the electrochemical output. In fact, an increase in anode catalyst loading from 1 to 4 mg cm−2 led to an increase in power density of 10%–30% for DMFCs, operating at the same conditions [25, 389, 390]. Considering DEFCs, moving from an anode catalyst loading of 2 to 4 mg cm−2, the increase in power density is less pronounced [25]. Despite a twofold or fourfold increase in the loading, a non-proportional increase in power output was recorded indicating that a positive effect is produced. However, not all the electrocatalyst fully utilized and an optimized electrocatalyst utilization should be pursued. Considering that in DAFCs operating with PGM-free cathode electrocatalysts, the anode Pt–Ru electrocatalyst counts as most of the MEA cost, and an increase in its loading led to an increase in the overall cost. Solutions might be pursued by tuning the anode catalyst layer and loading along the anodic flow field [391]. Therefore, a trade-off between performance and cost should be analyzed for each specific case and optimized.
Different cathode electrocatalysts, belonging to the family of PGM and PGM-free, have also been tested as reported above. Also in this case, the cathode electrocatalyst loading affects the power output. Generally, an increase in electrocatalyst loading led to an improved power output but typically with a trade-off. In fact, it was shown an increase in DMFC power output while increasing the cathode electrocatalyst loading (Fe–N–C electrocatalyst) from 2 to 4 mg cm−2 and finally to 6 mg cm−2 [392]. Similarly, in another study, it was shown an increase in power output by doubling the electrocatalyst layer from 2.5 to 5 mg cm−2 [393]. However, the benefit of a greater loading diminished with operating temperature moving from 30 to 90 °C, indicating that other factors might be influencing the DMFC performance. Another study on DMFCs has shown that the power density decreased when increasing the catalyst loading from 4 to 8 mg cm−2 [25]. This might be due to the thicker catalytic layer whose thickness increases importantly with the loading and leads to other parasitic losses such as mass transport issues for both reagents and products. An optimal cathode electrocatalyst loading should be found and correlated with the cathode catalyst layer thickness and porosity.
The cathode ionomer also plays a crucial role in the power output of DAFCs. It was found that among 35 wt%, 45 wt% and 55 wt% Nafion® ionomer used in the cathode electrocatalytic layer of an acid DMFC, 45 wt% was the most performing [392]. Similarly, an increase of Nafion® ionomer from 4 wt to 35 wt% and finally to 50 wt% led to an improvement in DMFC performance and then a decrease at the upper limit [394]. Consequently, an optimum ionomer content must be found to enhance the power output.
Also, the polymeric membrane affects importantly the power density. In hydrogen fuel cells, the general trend indicates that a decrease in the thickness of the membrane lowers the ohmic losses resulting in higher power density. This is not the case for DAFCs with PGM cathode electrocatalysts, where a thicker membrane is used to reduce the alcohol crossover transport phenomena and consequently mitigate the mixed potentials. Due to the high methanol tolerance of the PGM-free cathode electrocatalysts, a thinner membrane can be used successfully as shown very recently with a power density of 135 mW cm−2 obtained by using a Fe/Co–N–C cathode electrocatalyst operating at 80 °C under methanol-air conditions.
5.2 Effect of Operating Conditions on Power Output
Several operational parameters such as temperature, alcohol molarity, cathode airflow rates and water management have a critical role in the power output of DAFCs [395].
Operating temperature is an important parameter affecting the reaction kinetics of DAFCs and in turn the power output [25, 389, 396]. Generally, an increase in cell temperature leads to an increase in power output, independently from the electrolyte pH [25, 397]. However, limitations in the operations are given by the polymeric electrolytes that reduce their stability at higher conductivities. It was mentioned above that a narrow set of polymeric membranes (anion exchange or cation exchange depending on the selected environment) are used for low temperatures (below 100 °C). Moreover, as the temperature increases, the polymeric membranes (e.g., Nafion®) can suffer from increased ohmic resistance due to the loss of water molecules. At last, an increase of temperature can generally lead to an increase in fuel crossover phenomena which in turn lead to a lower utilization of fuel and therefore a lower Faradaic efficiency.
Another parameter affecting the power output of DAFCs is the alcohol molarity fed on the anode compartment for the oxidation reaction [25, 398, 399]. Due to the problems related to alcohol crossover, in the presence of PGM cathode electrocatalysts, a mixed potential is created that in turn lowers the OCV and the power output. Therefore, usually, the alcohol concentration fed to the anode is lower than the stoichiometric. As alcohol is diluted, an increasing concentration from 0.5 to 1 M and to 2 M leads to an increase in power output [25, 390]. However, further increase in molarity (> 2 M), has the effect of enhancing the mixed potential on the PGM cathode electrocatalyst lowering importantly the OCV and power output [25]. This is not necessary the case when PGM-free materials are used as electrocatalysts at the cathode. In fact, as PGM-free electrocatalysts do not interact with alcohol molecules, being defined as alcohol tolerant electrocatalysts, it was shown that an increase of methanol molarity up to 5 M led to an increase in power output [25, 390, 399]. Further increase in methanol molarity slightly lowers the power output. The increase in ethanol molarity instead, leads to a slight decrease in power output, but this can be ascribed to the sluggish anodic electrocatalysis rather than the cathodic one [25]. With the utilization of PGM-free electrocatalysts at the cathode, it was shown the possibility of operating at alcohol concentration up to 17 M, which is the stoichiometric ratio, without having high losses due to the alcohol crossover.
Another parameter affecting the power output of DAFCs is the reagent flow rate. It was shown that an increase in the liquid anodic flow rate can lead to an increase of the power output [400]. Particularly, despite OCV and activation overpotentials are not affected by the liquid anodic flow rate, limiting currents and concentration polarization might occur at earlier current densities. This phenomenon can be ascribed to the reaction occurring in the anode side creating a diphase flow. In fact, as methanol or ethanol mixtures entering the anode are completely liquid, the anodic reaction generates CO2 gas as final product meaning that with the gaseous bubble within the flow field, part of the electrode is not wet, meaning that is not directly involved in the reaction. The increase in the inlet flow rate would decrease the “dead zones” and in turn would decrease the residence time of the gas within the flow field. This leads to a more efficient utilization of the active electrodes and in turn higher power output would be produced.
Similarly, the airflow at the cathode can also affect importantly the power output of a DAFC. In fact, a greater air flow might lead to an increase in the pressure within the cathode flow field that in turn increase the oxygen partial pressure being beneficial for the power output. Moreover, a greater air flow might also remove the alcohol that moves from anode to cathode (crossover), “cleaning” the electrocatalyst active sites from the alcohol and reducing the mixed potentials. However, a high air flow might lead to dehydration of the membrane lowering its ion conductivity and in turn being detrimental for the power output. Therefore, a humidified airflow would be required increasing in turn the operational costs.
Water management is a key issue in all the low temperature fuel cells including DAFCs. An excessive presence of water on the cathode side might lead to flooding phenomena [381, 401,402,403,404,405] and lower the electrochemical performance. In parallel, the lack of water might dry the membrane and dramatically increase the membrane ion conductivity and in turn lower the performance. Therefore, an optimum has to be found in terms of anodic and cathodic feeding, temperature and utilization of GDLs. GDLs, often not considered critical, also have an important effect on the water management [406], methanol crossover [407] and finally power output [406].
5.3 Acidic versus Alkaline DAFCs
5.3.1 Performance of DMFCs in Acid Electrolytes
DMFCs with Pt/C electrocatalysts at the cathode exhibit peak power density (PPD, mW cm−2) ranging from 30 to 90 mW cm−2 [25, 408, 409] at temperatures up to 90 °C. The use of multiwall carbon nanotubes subjected to specific heat-treatment allowed to reach PPD up to 108 mW cm−2 [409]. In this specific case, Pt nanoparticles have been deposited on medium-unzipped multiwall carbon nanotubes (Pt/MU-MWCNT) with a tailored 3D nanoarchitecture which allowed to better exploit the performance of Pt towards ORR.
Considering the alcohol crossover phenomenon, bimetallic electrocatalysts are preferred. The use of Pt- and Pd-based alloys or bimetallic electrocatalysts (Pt–Pd, Pt–Ru, Pt–Ni, Pt–Co, Pt–Fe) was investigated with good success [410,411,412,413]. As an example, Pd–Pt bimetallic electrocatalysts highly rich in Pd demonstrated to be highly active and methanol tolerant in DMFCs, reaching a performance of 112 mW cm−2 when the amount of Pt is the lowest [410]. In fact, electrocatalysts with a higher Pt/Pd ratio showed a lower performance due to the high activity of Pt towards MOR. Evidence of DMFCs working at temperatures above 90 °C is also present in literature. Such devices showed a dramatic increase of power densities when cathodic Pt is alloyed with Ru (400 mW cm−2 using pure oxygen [216], 280 mW cm−2 using air [414]). Experimental evidence also suggested a strong influence of membrane materials on power density. The use of composite membranes could enhance power density output up to 400 mW cm−2 [415].
PGM-free electrocatalysts are very promising because of their high alcohol tolerance [381, 392, 416,417,418]. However, their ORR activity is relatively low due to the poor ORR kinetics in acidic medium, reaching PPD between 40 and 70 mW cm−2 at 60 °C. However, with PGM-free electrocatalysts, the MEA fabrication process plays a crucial role. Wang et al. [418] demonstrated that realizing a good triple-phase interface in the catalyst layer is necessary to boost the ORR activity. A good mixing of micropores, mesopores and macropores is necessary to assuring an optimal oxygen transport to reach the active sites, enhancing the electrode performance [419]. They demonstrated that with an engineered MEA with sufficient hydrophobicity and mass transport properties, the performance of a DMFC can increase by more than 40% on the maximum power peak density. They reached this result by introducing dimethyl silicon oil (DMS) in the electrode fabrication, increasing the performance of their Fe–N–C catalyst from 72 to 102 mW cm−2. Osmieri et al. [393] showed that a proper optimization of the electrocatalyst loading and the ionomer content in the electrocatalytic layer is also crucial to enhancing the DMFC performance by using a Fe–N–C electrocatalyst at the cathode. An engineered MEA can also prevent flooding of the cathode, enhancing the performance of the DMFC [381, 418, 420].
Bimetallic Fe–Co electrocatalysts can enhance DMFC performance as well. Shi et al. [421] reached a performance of 130 mW cm−2 (in air) with a dual-site Fe/Co–N–C electrocatalyst. The electrocatalyst was synthesized as a Co–N–C from a Co-doped ZIF-8 precursor with a controlled Zn/Co ratio, subsequently doped with Fe ions. A Zn/Co ratio equal to 11/2 was identified an optimal value. In fact, with an excessive Co-doping, the inactive Co-based metal species led to a promising ORR activity. The authors demonstrated that doping the basic Co–N–C catalyst with Fe allowed increasing the specific surface area and mesoporosity, which both favor oxygen diffusion to the active sites in the MEA.
An interesting approach to overcome the actual low performance of PGM-free electrocatalysts is the mixed approach adopted by Kosmala et al. [422]. A hybrid Pt/FeNC electrocatalyst, rich of FeNx sites and Pt@FeOx particles, showed excellent durability in acid media, together with a high tolerance to methanol. The physical–chemical characterization of the Pt/FeNC electrocatalyst containing only 2 wt% Pt showed the coexistence of Pt nanoparticles encapsulated by a thin Fe-oxide shell and FeNx sites. The core/shell structure of the electrocatalyst, where Pt is covered by a layer of Fe-oxides, makes this material inert to methanol oxidation and highly stable thanks to the stability of FeOx in acid environment. The high activity demonstrated by the hybrid materials can be attributed to the tunneling effect of Pt electrons through the FeOx ultrathin layer at the core/shell interface. In fact, electrons from the surface of Pt can tunnel through the Fe oxide thin layer and catalyze the oxygen reduction. Such an effect is present only when the thickness of FeOx surrounding the Pt nanoparticles is ultralow, otherwise the tunneling effect decreases progressively with the increase of thickness of the FeOx. Thus, the core/shell Pt/FeNC electrocatalysts can be envisaged as promising materials for stable PGM-free surfaces in acid media, and tolerant to methanol. Interestingly, these Pt@FeOx electrocatalysts could also be interesting HOR electrocatalysts for PEMFC anodes, thanks to their tolerance to species known to poison exposed Pt nanoparticles.
5.3.2 Performance of DEFCs in Acid Electrolytes
The use of ethanol in acid DAFCs is somehow limited because of the sluggish kinetics of the anodic EOR (involving C–C bond breaking) reaction compared to MOR, and of course the crossover of ethanol from the anode to cathode across the membrane. Similar to methanol, ethanol crossover results in parasitic EOR at the cathode, with the formation of a mixed potential and consequent lower fuel cell efficiency. With a Pt/C ORR electrocatalyst, the PPD does not overcome 9 mW cm−2 [25, 167] (Table 7). An ethanol-tolerant ORR electrocatalyst can mitigate the oxidation of ethanol at the cathode side. Few examples are available in the literature, where the most interesting electrocatalyst is the one described by Meenakshi et al. [423]. They developed a mixed Pt-TiO2/C electrocatalyst, using the so-called strong “d–d type” metal support interaction (SMSI) effect between Pt and TiO2. With this electrocatalyst, they obtained a PPD three times higher, reaching 31 mW cm−2 at 60 °C. Thanks to the SMSI effect, the Pt–H and Pt–CO bonds which limit the ORR are substantially weakening till suppression of intermediate formation. Transition metal oxides like TiO2 and WO3 are well-known for this effect. In fact, water molecules trapped inside the oxide network provide a hydrophilic behavior, which favors the OH− transfer within the system, avoiding the formation of intermediates, with consequent spillover of primary oxide over metallic electrocatalyst particles [424] and improvement of the ORR performance.
An example of acid DEFCs with PGM-free cathode electrocatalysts is reported by Sebastian et al. [25]. In this work, they compared MEAs with a Pt/C and a Fe–N–C electrocatalyst at the cathode. With the Fe–N–C catalyst, they reached a PPD of 12 mW cm−2 at 60 °C by feeding a 2 M ethanol solution (14 mW cm−2 at 90 °C with 5 M ethanol solution). They also performed a chronoamperometric test of 75 h to assess the stability of the PGM-free electrocatalyst. The MEA had a rapid decrease of performance in the initial 10 h of the experiment, with a current density loss rate of 3.5% per hour. Then, the current density loss was lower, stabilizing at a rate of about 0.2% per hour. Investigations on the reasons of the irreversible performance decay with time highlighted that it could be rather caused by the degradation of other MEA components, such as the anodic electrocatalyst for the alcohol oxidation catalyst (anode), or the ionomer or the membrane, rather than the cathodic electrocatalyst. It is well known that Ru present in the anodic electrocatalyst is susceptible to leaching out [425].
Figure 9 shows the morphology of some of the electrocatalysts listed in Table 7.

Morphology of some of the cathodic electrocatalysts for acid DMFCs listed in Table 7. a Scheme and TEM image of the unzipped Pt/MU-MWCNT. Adapted with permission from Ref. [409]. Copyright © 2019, American Chemical Society. b TEM image and EDS spectrum of the carbon-supported Pd19Pt1. Adapted with permission from Ref. [410]. Copyright © 2015, Elsevier Ltd. c TEM image of the Fe–Nx–C. Adapted with permission from Ref. [416]. Copyright © 2016, John Wiley & Sons, Inc. d STEM-EDXS images of the Pt2.0Fe1.0–N–C, with the highlight on a single Pt-rich nanoparticle. Adapted with permission from Ref. [422]. Copyright © 2020, American Chemical Society. e HAADF-STEM image and relative EELS analysis of the Fe/Co–N–C electrocatalyst, with Zn/Co = 11/2. Adapted with permission from Ref. [421]. Copyright © 2020, the Royal Society of Chemistry
5.3.3 Performance of DMFCs in Alkaline Electrolytes
Concerning DMFCs in alkaline environment with PGM-free cathode electrocatalysts the performance is relatively low, mostly because methanol oxidation leads to CO2 generation, which causes a rapid carbonation of the OH− exchange membrane. For example, Ratso et al. [426] did not overcome 7 mW cm−2 as PPD at 50 °C with Fe–N–C or Co–N–C electrocatalysts. A typical reference material for ORR in alkaline environment is MnO2, thanks to the effectiveness of the redox couple Mn3+/4+ [388, 427]. MEAs with a cathode composed of only MnO2 reached a PPD of 27 mW cm−2 at 40 °C [428]. The addition of M–N–C materials to MnO2 allowed reaching 41 mW cm−2 as PPD at 40 °C [428]. In this specific case, Fe–N–C electrocatalysts were prepared from urea–formaldehyde resins mixed with MnO2. Alkaline DMFCs displayed a superior ORR electrocatalytic activity thanks to the formation of Fe–Nx active sites and high amounts of pyridine-N. A chronoamperometric test showed an excellent durability with a minimal cell voltage decay after 39 h of testing.
Other examples in the literature show that a mix of oxides such as Fe2O3 and Mn2O3 (in the ratio 3:1) can reach a PPD of 46 mW cm−2 at 60 °C [429]. The good performance is ascribed to the formation of numerous heterojunctions between the two oxides, which form an intensive internal electric field favoring the electron transfer and thus the ORR. They demonstrated that the heterojunctions increased with the increase of the Fe/Mn ratio. In addition, the presence of Fe2O3, which is an n-type semiconductor, enhanced the oxygen storage capability of the electrocatalyst, favoring the Fe3+/2+ redox couple.
The highest performance reported so far in the literature has been reached with a hybrid electrocatalyst composed of three-dimensional Mn2O3 oxide deposited on ultrathin and porous nanosheets of Co1.2Ni1.8O4. With this electrocatalyst, Liu et al. [430] reached the remarkable PPD value of 120 mW cm−2 at 55 °C (70 mW cm−2 at 28 °C). The tolerance to methanol was very high, also at a concentration of 2 M. The durability of the performance was monitored by keeping the MEA at a constant current density of 30 mA cm−2 for 120 h. The voltage remained practically constant, showing almost no degradation effects. XRD analysis after the durability tests conducted on the cathodic electrocatalyst showed unchanged diffraction peaks, demonstrating the high stability of this hybrid electrocatalyst in alkaline environment. The excellent electrochemical activity of the Mn2O3@Co1.2Ni1.8O4 could be attributed to the narrow band gap of Co1.2Ni1.8O4, which accelerates the electrons jumping from the valence band to the conduction band. Consequently, Mn3+ is fast oxidized into Mn4+ providing an electron to the absorbed oxygen. Indeed, the excellent stability can be attributed to the high stability of Mn2O3 and to the partial substitution of Ni with Co in the Co1.2Ni1.8O4 mixed oxide.
Interestingly, Liu et al. [431] developed a highly performing and stable cathodic electrocatalyst based on NiC2O4. They obtained 151 mW cm−2 as PPD at 65 °C, compared to 18 mW cm−2 reached with a commercial Pt/C at the cathode side. Stability tests performed monitoring the voltage during galvanostatic test at 10 and 50 mA cm−2 at room temperature for 90 h demonstrated a very stable electrocatalyst, with limited cell voltage loss. The high activity towards ORR is provided by Co3+ surface ions, which act as functional active sites [432]. In fact, Co3+ in spinel metal oxides works as donor/acceptor reduction site. When Co3+ is oxidized to Co4+, it donates an excited electron to oxygen, which is reduced to OH− in a faster way compared to the reduction that can occur when the electron comes from the external circuit. The latter, instead, reaches easily the Co4+ ions to reduce them back to Co3+. Considering that the developed NiCo2O4 electrocatalyst has a high specific surface area thanks to its particular “desert-rose” morphology (Fig. 10), this electrocatalyst exhibited an extremely high activity. The authors concluded that Co3+ ions facilitate the direct electron capture by oxygen favoring thus an efficient reduction to OH−.

Morphology of some of the cathodic catalysts for alkaline DAFCs listed in Table 8. a TEM, HRTEM and EDS elemental mapping of Fe–N–C/MnO2. Adapted with permission from Ref. [428]. Copyright © 2019, Elsevier B.V. b SEM of Mn2O3, SEM, TEM, HRTEM, SAED patterns and EDX mapping of Mn2O3@Co1.2Ni1.8O4. Adapted with permission from Ref. [430]. Copyright © 2018, American Chemical Society. c FESEM and EDS elemental mapping of Fe2O3/Mn2O3 in the ratio 3:1. Adapted with permission from Ref. [429]. Copyright © 2018, the Royal Society of Chemistry. d SEM, TEM and HRTEM of NiCo2O4. Adapted with permission from Ref. [431]. Copyright © 2017, Elsevier B.V. e SEM of NiCo2O4. Adapted with permission from Ref. [433]. Copyright © 2019, American Chemical Society. f STEM and TEM of Fe–Co–N–C. Adapted with permission from Ref. [438]. Copyright © 2018, John Wiley & Sons, Inc
Tan et al. [433] also developed a Pt-free and Fe-free electrocatalyst based on NiCo2O4 at the cathode, able to reach 126 mW cm−2 as PPD (26 mW cm−2 when using air instead of pure oxygen). SEM analyses revealed that the morphology of NiCo2O4 has a series of porous nanosheets of about 1 μm thickness. This highly porous structure of the nanosheets shows a BET specific surface area of 54 m2 g−1 and pores with an average size of 17.6 nm. The MEA prepared with this NiCo2O4 electrocatalyst exhibited significant stability along a 200-h test at room temperature where the current density was kept constant at 50 mA cm−2. At the end of the durability test, the cell voltage dropped only by 20%.
5.3.4 Performance of DEFCs in Alkaline Electrolytes
Pt-based electrocatalysts are generally less effective in alkaline than in acidic conditions because of the limited activity of Pt towards ORR in alkaline media [434]. The best activity reached with a Pt/C at the cathode has been recorded by Fujiwara et al. [167], who obtained 58 mW cm−2 as PPD at room temperature, feeding air.
MnO2-based electrocatalysts at the cathode have been used also for alkaline DEFCs, but the performance was low. For example, Gaurava et al. [435] reached 52 mW cm−2 as PPD at 45 °C with a 4 MEA simple stack fed with air.
Promising results have been achieved also by using metal oxides as perovskites [436]. With a cathode composed of La0.7Sr0.3MnO3 perovskite it was possible to reach 28 mW cm−2 as PPD at 30 °C. In-situ half-cell measurements demonstrated the high tolerance of this perovskite toward ethanol.
Higher performance has been achieved with M–N–C cathode electrocatalysts, due to enhanced ORR kinetics in alkaline media and extraordinary tolerance to alcohols [437]. With Fe–N–C obtained from a mesoporous carbon derived by sacrificial templating of mesoporous silica, the maximum PPD recorded was 72 mW cm−2 at 90 °C. Accelerated stability tests showed a remarkable performance decay after few hours of operation, with ca. 45% loss in the peak power density. Interestingly, the initial performance was fully recovered after a purging/drying procedure. This procedure consisted in flowing dry nitrogen at 90 °C for half an hour. The purging procedure demonstrated that the performance loss was mainly due to flooding problems (too high-water presence within the cathode catalytic layer) rather than deactivation of the electrocatalyst.
With a similar sacrificial template procedure, Osmieri et al. [394] synthesized a series of bimetallic FeCo–N–C electrocatalysts, which reached 28 mW cm−2 as PPD in a passive DEFC at room temperature with important stability. In this case, the activity was most likely due to the presence of Co–Fe@C nanoparticles. Figure 10 shows the morphology of some of the electrocatalysts listed in Table 8.
5.4 Energy Efficiency: Dependency on Acidic Versus Alkaline Working Environment
Energy efficiency data for DAFCs are rather scarce in literature despite being a crucial parameter to be considered. In Table 9, some examples related to alkaline and acidic media for ethanol and methanol are reported identifying studies targeting the determination of energy efficiency; for the sake of comparison, the potential efficiency (estimated from the power curve) of some state-of-the-art examples is reported as well. The potential efficiency is the ratio of the cell potential to the theoretical thermodynamic potential for the reaction. The conversion is defined as the moles of fuel consumed compared to the total moles fed; this affects also Faradaic efficiency that takes also into account the selectivity of the oxidation in terms of number of electrons transferred. The main factor limiting the efficiency of the systems is the low fuel rate, since the decrease of the concentration hinders the mass transport generating potential losses that decrease the potential efficiency. Another limiting factor (for ethanol) is the incomplete oxidation of the alcohol due to the inability of cleaving the C–C (in particular in alkaline media), producing less charge for one mole of fuel, reducing the Faradaic efficiency and, in turn, the total energy efficiency. For example, ethanol when oxidized to acetic acid delivers 4 electrons instead of 12 for each molecule, reducing to 33% the maximum energy efficiency. Methanol does not suffer this problem as the oxidation selectively produces carbon dioxide (or carbonate). Due to these drawbacks, the reported overall efficiencies are rather low. The highest energy efficiency for ethanol in alkaline media was reported by Wang et al. [439] for Pd-(CeO2)/C anode electrocatalyst with a conversion from 46% to acetic acid (4 electrons) and a Faradaic efficiency of 15% with an overall energy efficiency (fuel to electrical energy) of 6.5% (the product energy content was not considered as gain). Similar results are reported by Bianchini et al. with a nanostructured Pd–NiZn/C anodic electrocatalyst and a Fe–Co/C cathodic electrocatalyst in alkaline media [168]. In acidic media instead, the highest reported energy efficiency for methanol is 25%, by Seo et al. that studied extensively the methanol crossover losses in various experimental conditions [440]. Another noteworthy study by Jiang et. al reported a conversion up to 86% for methanol with a peak power density of 26 mW cm−2 but a lower overall efficiency of 17% [441]. These studies used Pt-Ru alloy at the anode and Pt at the cathode with a Nafion® 117 membrane as electrolyte [440,441,442,443].
6 Market Perspectives
Up to now, the DMFC market includes portable, stationary and transportation devices, with portable applications covering the largest share (approx. 64%), followed by transportation (25%) and stationary (11%), as back-up power systems. For portable applications, DMFCs offer the most feasible option thanks to their high energy density and fast recharging options, compactness and lightweight. Methanol, in fact, can be easily stored, supplied and handled, sometimes also mixed with water (50/50 or 60/40 methanol/water).
The main players in DMFC companies are mostly located in North America (Ensol System in Canada, MeOH Power in the U.S.A.), North Europe (SFC Energy AG in Germany, Advent Technologies A/S in Denmark), and Asia Pacific (Samsung SDI Co. Ltd. in the Republic of Korea; Toshiba Corporation, Fujikura Ltd. and Panasonic Corporation in Japan). Table 10 provides an overview of the main DMFC commercial products available on the web.
Considering the major drawback and limitations of DMFCs, the global market size is limited compared to other power source systems such as batteries. In 2020, the DMFC market size reached 1.73 million USD, with an expected growth to 5.85 million USD in 2028, equivalent to a compound annual growth rate (CAGR) of 16.6% [452]. In Europe, the company SFC Energy AG (Germany) declared in its Quarterly Release Q3/2022 almost 44 million EUR of sales as “Clean Energy” products (hydrogen/methanol fuel cells and integrated products) in the period January–September 2022, with an increase of + 45% compared to the same period in 2021 [453]. The largest contribution to sales was determined predominantly by fuel cell solutions for industrial applications (almost doubled compared to the same period of the previous year), while sales to customers for private or recreational use decreased remarkably. For SFC Energy AG, the breakdown of sales by region has Europe as the top buyer (more than 47% of the market share), followed by the United States (43%), Asia (9%), and the rest of the world. The even more pressing request of clean power sources, combined with the availability and ease of handling of methanol will facilitate the market penetration of DMFCs. In fact, DMFCs, can provide high energy conversion together with flexible and clean operation when direct electrification and batteries cannot cover the request. Considering the 2030 Climate Target Plan [454] issued by the European Commission following the Paris Agreement (reducing greenhouse gas emissions to at least 55% below 1990’ levels by 2030 and achieving climate neutrality by 2050), the forecast sales for fuel cells are expected to increase by 2023 notwithstanding the global economy has become increasingly depressing because of the unstable geo-political circumstances, increasing inflation and unpredictable COVID policy around the world.
7 Problems to Overcome and Future Outlook
Actual DAFCs still suffer from limitations mainly related to the activity of the anode electrocatalysts. So far acidic DMFCs are the most developed systems and have reached the commercialization stage. In 2019, a logistic vehicle powered with DMFCs has been launched in China [459] and DMFCs have also been proposed as range extenders for battery vehicles. However, a future of DMFCs in high power applications is still hard to imagine for the concurrency of hydrogen systems, even on the light of the significant critical raw materials loading utilized. In almost all the cases, Pt loading usually exceeds 1 mg cm−2, and more often, to guarantee more stability and performance, 3 mg cm−2 Pt loadings are used with many examples in the literature that sometimes even exceed 5 mg cm−2. Acidic ethanol systems are less developed compared to methanol. Still, most of the reported works use large PGMs loading. The most relevant challenge in DEFCs is the conversion to CO2 that is still a minor oxidation product and that leads to low energy conversion efficiencies. Despite a large research effort most relevant electrocatalysts are far from achieving a complete conversion. Stability is also an issue and the targets of hydrogen PEMFCs are still far to be achieved. The reasons for the performance drops are various. In acidic system, the problem of cathode flooding generates a significant drop in the performance. Moreover, anode electrocatalysts suffer from stability issues. For the oxidation of methanol and ethanol, the most developed electrocatalysts are Pt–Ru alloys (50%/50% at.). These systems are prone to Ru leaching that has been demonstrated even in the first 20 h. of operation. The depletion of the Ru content at the surface of the electrocatalysts is a significant problem as Ru has the role to reduce CO poisoning and in turn improve durability.
A further issue that affects the acidic systems is alcohol crossover with the onset of mixed potential that determines loss in cell potential and ultimately in energy efficiency. This problem is mostly related to the use of cathode electrocatalysts that beside being efficient for the ORR are also effective for alcohols oxidation. The use of PGM-free electrocatalysts such as single atoms iron and cobalt catalysts is the most promising strategy to mitigate the problem of crossover, together with the development of new membranes that show less permeability to alcohols. The problem is less relevant in alkaline DEFCs because in alkaline conditions PGM-free electrocatalysts are more effective and show larger stability.
A major drawback seldom reported of alkaline systems is the use of a fuel mixed with concentrated alkali. Oppositely, acidic system exploits the acidity of the membrane only, allowing the use of fuels consisting of alcohols in deionized water solutions. The reason why alkaline systems require the use of concentrated alkali resides in the stoichiometry of the reaction. Indeed, the oxidation of alcohols in alkaline environment leads to the formation of anions (e.g., carbonate and acetate) as products. The rightmost negative charge is balanced by the stoichiometric consumption of the OH− at the reagent side. This fact combines with the kinetics of the oxidation reaction that, e.g., in the case of ethanol is dominated by the adsorption of OH− at the surface of the electrocatalyst at least at low overpotential (anode potential lower than 0.5 V RHE), a region that is extremely important for the fuel cells. The effect of fuel depletion has been seldom studied in the literature, but investigations shows that when the concentration of OH− in the fuel drops below 1 M the performance significantly decreases. This aspect must be considered while engineering systems to maximize the efficiency in the fuel utilization. The need to use OH− also affects the energetics of the whole system, as the production of alkali, usually by the chloro-soda industry, is very energy intensive and can give a significant contribution to the energy balance of the direct alcohol fuel cells. This aspect has already been shown in the analysis of the electrochemical reforming of ethanol [28, 29]. However, this point has never been stressed for the DAFCs. We believe that the implication of such facts is that when evaluating the energy input of alkaline DAFCs the energy content of the OH− should be considered in addition to that of the alcohols. This is a direct consequence of the stoichiometric OH− consumption that leads to the final conclusion that the OH− shall be considered as a component of the fuel and not just as a tool to modify the pH of the anode compartment.
The use of alkaline electrolytes however has also positive implications. Indeed, the anode is immerged in a strongly conductive electrolyte. This allows the use of electrodes consisting of thick layers of easy-to-handle materials as reported in [7]. These systems, thanks to an extremely high mass transport have achieved power density over 300 mW cm−2 in the case of DEFCs, even if using Pd loading up to 8 mg cm−2 with current densities largely exceeding 1 A cm−2. Remarkably, the anode catalyst layer engineering in alkaline systems could be much different from that of acidic systems, as the ion transport can be provided by the electrolyte itself whitout the need of ionomers and binders. The catalyst layers consist (e.g., titania nanotube arrays deposited onto titanium non-woven web [35, 460, 461]).
A further issue related to the alkaline system is safety. High performance systems need indeed highly concentrated alkali that must be handled with care during refueling. Moreover, eventual leakage of the systems may be dangerous for the devices and for the safety of the operator. This is not an issue for acid systems whereas mentioned above the fuel only consists of a solution of alcohols in water.
Fuel composition is also an issue. Indeed, neither in acidic nor in alkali can alcohols be used in pure forms. Diluted concentrations must be used to guarantee the best performances.
In alkaline systems most of the experiments have dealt with 1 M or 2 M ethanol solution. This means that the energy density of the fuel is limited by the low concentration of the alcohol. Moreover, the weight energy density is affected by the need to load the system also with hydroxide. The need to load alkaline DEFCs with alkali also affects the energy density of the fuel. Indeed, the addition of NaOH equimolar to e.g. ethanol, approximatively halves the energy density of the fuel.
Also, in acidic systems, due to alcohol crossover constraints, fuel concentrations are ranging between 1 and 2 M, reaching in some case 5 M solutions, when innovative (mainly composite) membranes with lower fuel permeability are used. Only few examples, using Fe-N-C electrocatalysts at the cathode, have demonstrated the feasibility of DAFC operation with high fuel concentration.
All these aspects suggest that while the high energy density of alcohols as fuels have been largely advocated as an advantage, translating it into practical applications is completely a different matter spotting on the light several important issues. Kinetics imposes severe constraints on the concentration of the alcohols in the fuel and the need to have them diluted in water decreases much the volumetric and the weight energy density of the devices. In alkaline systems the need of the equivalents of alkali makes the situation even worse.
For all what said above, there is the need for substantial multidisciplinary research to improve the performance of the devices. In the opinion of the authors, the reduction of PGM electrocatalyst loadings and the increase of the alcohol concentration in the fuel are among the most relevant issues demanding a substantial research effort. Secondarily, there is the need to improve the energetics of the anodic processes. Anode performances are indeed the most relevant in terms of potential drops and electrocatalysts with improved kinetics would improve the performance. Membranes also deserve much attention. Indeed, crossover is a limiting factor for the performance and the efficiency of the devices and depends much on the membrane. The problems interrelate with the concentration of the alcohol in the fuel; higher concentrations of alcohol in the fuel result in higher crossover. The limitation of crossover can come from leap in the membrane technology through the use of composite materials.
References
Lavacchi, A., Miller, H., Vizza, F.: Nanotechnology in Electrocatalysis for Energy. Springer, New York (2013). https://doi.org/10.1007/978-1-4899-8059-5
European Commission: Hydrogen and fuel cells. https://joint-research-centre.ec.europa.eu/scientific-activities-z/hydrogen-and-fuel-cells_en
Sproat, V., LaHurd, D.: Fuel cell balance-of-plant reliability testbed project. OSTI.GOV (2016). https://doi.org/10.2172/1335164
Akay, R.G., Yurtcan, A.B. (eds.): Direct Liquid Fuel Cells. Elsevier, Amsterdam (2021). https://doi.org/10.1016/c2018-0-04168-7
Coutanceau, C., Baranton, S.: Electrochemical conversion of alcohols for hydrogen production: a short overview. Wiley Interdiscip. Rev. Energy Environ. 5, 388–400 (2016). https://doi.org/10.1002/wene.193
Wang, D., Wang, P., Wang, S.C., et al.: Direct electrochemical oxidation of alcohols with hydrogen evolution in continuous-flow reactor. Nat. Commun. 10, 2796 (2019). https://doi.org/10.1038/s41467-019-10928-0
Chen, Y., Bellini, M., Bevilacqua, M., et al.: Direct alcohol fuel cells: toward the power densities of hydrogen-fed proton exchange membrane fuel cells. Chemsuschem 8, 524–533 (2015). https://doi.org/10.1002/cssc.201402999
U.S. Department of Energy: Alternative fuels data center. https://afdc.energy.gov/fuels/ethanol.html
Yun, Y.: Alcohol fuels: current status and future direction. In: Alcohol Fuels: Current Technologies and Future Prospect. IntechOpen (2020). https://doi.org/10.5772/intechopen.89788
Heat of combustion. Wikipedia Page. https://en.wikipedia.org/wiki/Heat_of_combustion
Heat of combustion. Hydrogen Tools. https://aws-beta.h2tools.org/hyarc/calculator-tools/lower-and-higher-heating-values-fuels
Ghumman, A., Pickup, P.G.: Efficient electrochemical oxidation of ethanol to carbon dioxide in a fuel cell at ambient temperature. J. Power Sources 179, 280–285 (2008). https://doi.org/10.1016/j.jpowsour.2007.12.071
Kowal, A., Li, M., Shao, M., et al.: Ternary Pt/Rh/SnO2 electrocatalysts for oxidizing ethanol to CO2. Nat. Mater. 8, 325–330 (2009). https://doi.org/10.1038/nmat2359
Chang, J.F., Wang, G.Z., Wang, M.Y., et al.: Improving Pd–N–C fuel cell electrocatalysts through fluorination-driven rearrangements of local coordination environment. Nat. Energy 6, 1144–1153 (2021). https://doi.org/10.1038/s41560-021-00940-4
Liu, F.Q., Wang, C.Y.: Mixed potential in a direct methanol fuel cell. J. Electrochem. Soc. 154, B514 (2007). https://doi.org/10.1149/1.2718404
Pan, M.Z., Pan, C.J., Li, C., et al.: A review of membranes in proton exchange membrane fuel cells: transport phenomena, performance and durability. Renew. Sustain. Energy Rev. 141, 110771 (2021). https://doi.org/10.1016/j.rser.2021.110771
You, W., Noonan, K.J.T., Coates, G.W.: Alkaline-stable anion exchange membranes: a review of synthetic approaches. Prog. Polym. Sci. 100, 101177 (2020). https://doi.org/10.1016/j.progpolymsci.2019.101177
Wang, L.Q., Bambagioni, V., Bevilacqua, M., et al.: Sodium borohydride as an additive to enhance the performance of direct ethanol fuel cells. J. Power Sources 195, 8036–8043 (2010). https://doi.org/10.1016/j.jpowsour.2010.06.101
Wang, L.Q., Lavacchi, A., Bellini, M., et al.: Deactivation of palladium electrocatalysts for alcohols oxidation in basic electrolytes. Electrochim. Acta 177, 100–106 (2015). https://doi.org/10.1016/j.electacta.2015.02.026
Modibedi, R.M., Ozoemena, K.I., Mathe, M.K.: Palladium-based nanocatalysts for alcohol electrooxidation in alkaline media. In: Shao, M. (ed.) Electrocatalysis in Fuel Cells, vol. 9, pp. 129–156. Springer, London (2013). https://doi.org/10.1007/978-1-4471-4911-8_6
Papadias, D.D., Ahluwalia, R.K., Thomson, J.K., et al.: Degradation of SS316L bipolar plates in simulated fuel cell environment: corrosion rate, barrier film formation kinetics and contact resistance. J. Power Sources 273, 1237–1249 (2015). https://doi.org/10.1016/j.jpowsour.2014.02.053
Hren, M., Božič, M., Fakin, D., et al.: Alkaline membrane fuel cells: anion exchange membranes and fuels. Sustain. Energy Fuels 5, 604–637 (2021). https://doi.org/10.1039/d0se01373k
Rizo, R., Arán-Ais, R.M., Padgett, E., et al.: Pt-richcore/Sn-richsubsurface/Ptskin nanocubes as highly active and stable electrocatalysts for the ethanol oxidation reaction. J. Am. Chem. Soc. 140, 3791–3797 (2018). https://doi.org/10.1021/jacs.8b00588
Sebastián, D., Baglio, V., Aricò, A.S., et al.: Performance analysis of a non-platinum group metal catalyst based on iron-aminoantipyrine for direct methanol fuel cells. Appl. Catal. B Environ. 182, 297–305 (2016). https://doi.org/10.1016/j.apcatb.2015.09.043
Sebastián, D., Serov, A., Matanovic, I., et al.: Insights on the extraordinary tolerance to alcohols of Fe–N–C cathode catalysts in highly performing direct alcohol fuel cells. Nano Energy 34, 195–204 (2017). https://doi.org/10.1016/j.nanoen.2017.02.039
Batista, B.C., Sitta, E., Eiswirth, M., et al.: Autocatalysis in the open circuit interaction of alcohol molecules with oxidized Pt surfaces. Phys. Chem. Chem. Phys. 10, 6686 (2008). https://doi.org/10.1039/b811787j
Liang, Z.X., Zhao, T.S., Xu, J.B., et al.: Mechanism study of the ethanol oxidation reaction on palladium in alkaline media. Electrochim. Acta 54, 2203–2208 (2009). https://doi.org/10.1016/j.electacta.2008.10.034
Miller, H.A., Lavacchi, A., Vizza, F.: Storage of renewable energy in fuels and chemicals through electrochemical reforming of bioalcohols. Curr. Opin. Electrochem. 21, 140–145 (2020). https://doi.org/10.1016/j.coelec.2020.02.001
Chen, Y.X., Lavacchi, A., Miller, H.A., et al.: Nanotechnology makes biomass electrolysis more energy efficient than water electrolysis. Nat. Commun. 5, 4036 (2014). https://doi.org/10.1038/ncomms5036
Pinto, A.M.F.R., Oliveira, V.B., Falcão, D.S.: Introduction to direct alcohol fuel cells. In: Pinto, A.M.F.R., Oliveira, V.B., Falcão, D.S. (eds.) Direct Alcohol Fuel Cells for Portable Applications, pp. 1–15. Elsevier, Amsterdam (2018). https://doi.org/10.1016/b978-0-12-811849-8.00001-2
Berretti, E., Longhi, M., Atanassov, P., et al.: Platinum group metal-free Fe-based (FeNC) oxygen reduction electrocatalysts for direct alcohol fuel cells. Curr. Opin. Electrochem. 29, 100756 (2021). https://doi.org/10.1016/j.coelec.2021.100756
Mathur, V.K., Crawford, J.: Fundamentals of gas diffusion layers in PEM fuel cells. In: Basu, S. (ed.) Recent Trends in Fuel Cell Science and Technology, pp. 116–128. Springer, New York (2007). https://doi.org/10.1007/978-0-387-68815-2_4
Ali Abdelkareem, M., Sayed, E.T., Nakagawa, N.: Significance of diffusion layers on the performance of liquid and vapor feed passive direct methanol fuel cells. Energy 209, 118492 (2020). https://doi.org/10.1016/j.energy.2020.118492
Arisetty, S., Prasad, A.K., Advani, S.G.: Metal foams as flow field and gas diffusion layer in direct methanol fuel cells. J. Power Sources 165, 49–57 (2007). https://doi.org/10.1016/j.jpowsour.2006.12.008
Lavacchi, A., Bellini, M., Berretti, E., et al.: Titanium dioxide nanomaterials in electrocatalysis for energy. Curr. Opin. Electrochem. 28, 100720 (2021). https://doi.org/10.1016/j.coelec.2021.100720
Fadzillah, D.M., Kamarudin, S.K., Zainoodin, M.A., et al.: Critical challenges in the system development of direct alcohol fuel cells as portable power supplies: an overview. Int. J. Hydrog. Energy 44, 3031–3054 (2019). https://doi.org/10.1016/j.ijhydene.2018.11.089
Petrii, O.A.: The progress in understanding the mechanisms of methanol and formic acid electrooxidation on platinum group metals (a review). Russ. J. Electrochem. 55, 1–33 (2019). https://doi.org/10.1134/s1023193519010129
Zoski, C.G. (ed.): Handbook of Electrochemistry. Elsevier, Amsterdam (2007). https://doi.org/10.1016/b978-0-444-51958-0.x5000-9
Riyanto, R., Othman, M.R., Salimon, J.: Synthesis of acetic acid from ethanol by electrooxidation technique using Ni–Cu–PVC electrode. ASEAN J. Sci. Technol. Dev. 25, 363–371 (2017). https://doi.org/10.29037/ajstd.267
Chen, Z.W., Dodelet, J.P., Zhang Dodelet, J.J. (eds.): Non-noble Metal Fuel Cell Catalysts. Wiley, Weinheim (2014). https://doi.org/10.1002/9783527664900
Bevilacqua, M., Filippi, J., Lavacchi, A., et al.: Energy savings in the conversion of CO2 to fuels using an electrolytic device. Energy Technol. 2, 522–525 (2014). https://doi.org/10.1002/ente.201402014
Stuve, E.M.: Overpotentials in electrochemical cells. In: Kreysa, G., Ota, K., Savinell, R.F. (eds.) Encyclopedia of Applied Electrochemistry, pp. 1445–1453. Springer, New York (2014). https://doi.org/10.1007/978-1-4419-6996-5_330
Léger, J.-M.: Mechanistic aspects of methanol oxidation on platinum-based electrocatalysts. J. Appl. Electrochem. 31, 767–771 (2001). https://doi.org/10.1023/A:1017531225171
Gasteiger, H.A., Markovic, N., Ross, P.N., Jr., et al.: Methanol electrooxidation on well-characterized platinum-ruthenium bulk alloys. J. Phys. Chem. 97, 12020–12029 (1993). https://doi.org/10.1021/j100148a030
Lamy, C., Lima, A., LeRhun, V., et al.: Recent advances in the development of direct alcohol fuel cells (DAFC). J. Power Sources 105, 283–296 (2002). https://doi.org/10.1016/S0378-7753(01)00954-5
Fang, X., Wang, L.Q., Shen, P.K., et al.: An in situ Fourier transform infrared spectroelectrochemical study on ethanol electrooxidation on Pd in alkaline solution. J. Power Sources 195, 1375–1378 (2010). https://doi.org/10.1016/j.jpowsour.2009.09.025
Bevilacqua, M., Bianchini, C., Marchionni, A., et al.: Improvement in the efficiency of an OrganoMetallic Fuel Cell by tuning the molecular architecture of the anode electrocatalyst and the nature of the carbon support. Energy Environ. Sci. 5, 8608–8620 (2012). https://doi.org/10.1039/C2EE22055E
Rus, E.D., Wakabayashi, R.H., Wang, H.S., et al.: Methanol oxidation at platinum in alkaline media: a study of the effects of hydroxide concentration and of mass transport. ChemPhysChem 22, 1397–1406 (2021). https://doi.org/10.1002/cphc.202100087
Yaqoob, L., Noor, T., Iqbal, N.: A comprehensive and critical review of the recent progress in electrocatalysts for the ethanol oxidation reaction. RSC Adv. 11, 16768–16804 (2021). https://doi.org/10.1039/d1ra01841h
Bambagioni, V., Bianchini, C., Chen, Y.X., et al.: Energy efficiency enhancement of ethanol electrooxidation on Pd–CeO2/C in passive and active polymer electrolyte-membrane fuel cells. Chemsuschem 5, 1266–1273 (2012). https://doi.org/10.1002/cssc.201100738
Scott, K., Xing, L.: Direct methanol fuel cells. In: Sundmacher, K. (ed.) Fuel Cell Engineering, vol. 41, pp. 145–196. Elsevier, Amsterdam (2012). https://doi.org/10.1016/b978-0-12-386874-9.00005-1
Pineri, M., Eisenberg, A. (eds.): Structure and Properties of Ionomers. Springer, Dordrecht (1987). https://doi.org/10.1007/978-94-009-3829-8
Samms, S.R., Wasmus, S., Savinell, R.F.: Thermal stability of Nafion® in simulated fuel cell environments. J. Electrochem. Soc. 143, 1498–1504 (1996). https://doi.org/10.1149/1.1836669
Brandão, L., Rodrigues, J., Madeira, L.M., et al.: Methanol crossover reduction by Nafion modification with palladium composite nanoparticles: application to direct methanol fuel cells. Int. J. Hydrog. Energy 35, 11561–11567 (2010). https://doi.org/10.1016/j.ijhydene.2010.04.096
Ball Reviewed by Sarah C: Electrochemistry of proton conducting membrane fuel cells. Platin. Metals Rev. 49, 27–32 (2005). https://doi.org/10.1595/147106705x25525
Ling, J., Savadogo, O.: Comparison of methanol crossover among four types of Nafion membranes. J. Electrochem. Soc. 151, A1604 (2004). https://doi.org/10.1149/1.1789394
Miyake, T.O. N., Wakizoe, M., Honda, E.: Proceedings of the fourth international symposium on proton conducting membrane fuel cells. In: Proceedings of the Fourth International Symposium on Proton Conducting Membrane Fuel Cells, p. W-1880 (2004)
Arico, A.S., Baglio, V., Di Blasi, A., et al.: Proton exchange membranes based on the short-side-chain perfluorinated ionomer for high temperature direct methanol fuel cells. Desalination 199, 271–273 (2006). https://doi.org/10.1016/j.desal.2006.03.065
Yoshitake, E.Y.M., Kunisa, Y., Endoh, E.: Solid polymer type fuel cell and production method thereof. US Patent 6,933,071, 23 Aug 2005
Penner, R.M., Martin, C.R.: Ion transporting composite membranes. I. Nafion-impregnated Gore-Tex. J. Electrochem. Soc. 132, 514–515 (1985). https://doi.org/10.1149/1.2113875
Bahar, B., Hobson, A.R., Kolde, J.A., et al.: Ultra-thin integral composite membrane. US Patent 5,547,551, 20 Aug 1996
Savadogo, O.: Emerging membranes for electrochemical systems: (I) solid polymer electrolyte membranes for fuel cell systems. ChemInform (1998). https://doi.org/10.1002/chin.199847334
Laberty-Robert, C., Vallé, K., Pereira, F., et al.: Design and properties of functional hybrid organic-inorganic membranes for fuel cells. Chem. Soc. Rev. 40, 961–1005 (2011). https://doi.org/10.1039/C0CS00144A
Mu, S.C., Tang, H.L., Wan, Z.H., et al.: Au nanoparticles self-assembled onto Nafion membranes for use as methanol-blocking barriers. Electrochem. Commun. 7, 1143–1147 (2005). https://doi.org/10.1016/j.elecom.2005.08.019
Yoon, S.R., Hwang, G.H., Cho, W.I., et al.: Modification of polymer electrolyte membranes for DMFCs using Pd films formed by sputtering. J. Power Sources 106, 215–223 (2002). https://doi.org/10.1016/S0378-7753(01)01048-5
Ma, Z.: A palladium-alloy deposited Nafion membrane for direct methanol fuel cells. J. Membr. Sci. 215, 327–336 (2003). https://doi.org/10.1016/s0376-7388(03)00026-7
Kim, Y.M., Park, K.W., Choi, J.H., et al.: A Pd-impregnated nanocomposite Nafion membrane for use in high-concentration methanol fuel in DMFC. Electrochem. Commun. 5, 571–574 (2003). https://doi.org/10.1016/S1388-2481(03)00130-9
Tang, H.L., Pan, M., Jiang, S.P., et al.: Self-assembling multi-layer Pd nanoparticles onto Nafion™ membrane to reduce methanol crossover. Colloids Surf. A Physicochem. Eng. Asp. 262, 65–70 (2005). https://doi.org/10.1016/j.colsurfa.2005.04.011
Liang, Z.X., Shi, J.Y., Liao, S.J., et al.: Noble metal nanowires incorporated Nafion® membranes for reduction of methanol crossover in direct methanol fuel cells. Int. J. Hydrog. Energy 35, 9182–9185 (2010). https://doi.org/10.1016/j.ijhydene.2010.06.054
Jiang, S.P., Liu, Z.C., Tang, H.L., et al.: Synthesis and characterization of PDDA-stabilized Pt nanoparticles for direct methanol fuel cells. Electrochim. Acta 51, 5721–5730 (2006). https://doi.org/10.1016/j.electacta.2006.03.006
Jung, E.H., Jung, U.H., Yang, T.H., et al.: Methanol crossover through PtRu/Nafion composite membrane for a direct methanol fuel cell. Int. J. Hydrog. Energy 32, 903–907 (2007). https://doi.org/10.1016/j.ijhydene.2006.12.014
Prabhuram, J., Zhao, T.S., Liang, Z.X., et al.: Pd and Pd–Cu alloy deposited Nafion membranes for reduction of methanol crossover in direct methanol fuel cells. J. Electrochem. Soc. 152, A1390 (2005). https://doi.org/10.1149/1.1926671
Iwai, Y., Ikemoto, S., Haramaki, K., et al.: Influence of ligands of palladium complexes on palladium/Nafion composite membranes for direct methanol fuel cells by supercritical CO2 impregnation method. J. Supercrit. Fluids 94, 48–58 (2014). https://doi.org/10.1016/j.supflu.2014.06.015
Thiam, H.S., Daud, W.R.W., Kamarudin, S.K., et al.: Performance of direct methanol fuel cell with a palladium-silica nanofibre/Nafion composite membrane. Energy Convers. Manag. 75, 718–726 (2013). https://doi.org/10.1016/j.enconman.2013.08.009
Kim, Y., Choi, W., Woo, S., et al.: Proton conductivity and methanol permeation in Nafion™/ORMOSIL prepared with various organic silanes. J. Membr. Sci. 238, 213–222 (2004). https://doi.org/10.1016/j.memsci.2004.04.005
Li, C.N., Sun, G.Q., Ren, S.Z., et al.: Casting Nafion-sulfonated organosilica nano-composite membranes used in direct methanol fuel cells. J. Membr. Sci. 272, 50–57 (2006). https://doi.org/10.1016/j.memsci.2005.07.032
Ren, S.Z., Sun, G.Q., Li, C.N., et al.: Organic silica/Nafion® composite membrane for direct methanol fuel cells. J. Membr. Sci. 282, 450–455 (2006). https://doi.org/10.1016/j.memsci.2006.05.050
Liang, Z.X., Zhao, T.S., Prabhuram, J.: Diphenylsilicate-incorporated Nafion® membranes for reduction of methanol crossover in direct methanol fuel cells. J. Membr. Sci. 283, 219–224 (2006). https://doi.org/10.1016/j.memsci.2006.06.031
Tay, S.W., Zhang, X.H., Liu, Z.L., et al.: Composite Nafion® membrane embedded with hybrid nanofillers for promoting direct methanol fuel cell performance. J. Membr. Sci. 321, 139–145 (2008). https://doi.org/10.1016/j.memsci.2008.04.049
Li, T., Yang, Y.: A novel inorganic/organic composite membrane tailored by various organic silane coupling agents for use in direct methanol fuel cells. J. Power Sources 187, 332–340 (2009). https://doi.org/10.1016/j.jpowsour.2008.11.035
Sahu, A.K., Meenakshi, S., Bhat, S.D., et al.: Meso-structured silica-Nafion hybrid membranes for direct methanol fuel cells. J. Electrochem. Soc. 159, F702–F710 (2012). https://doi.org/10.1149/2.036211jes
Tricoli, V., Nannetti, F.: Zeolite-Nafion composites as ion conducting membrane materials. Electrochim. Acta 48, 2625–2633 (2003). https://doi.org/10.1016/S0013-4686(03)00306-2
Baglio, V., Arico, A.S., Di Blasi, A., et al.: Zeolite-based composite membranes for high temperature direct methanol fuel cells. J. Appl. Electrochem. 35, 207–212 (2005). https://doi.org/10.1007/s10800-004-6202-z
Baglio, V., Di Blasi, A., Aricò, A.S., et al.: Investigation of the electrochemical behaviour in DMFCs of chabazite and clinoptilolite-based composite membranes. Electrochim. Acta 50, 5181–5188 (2005). https://doi.org/10.1016/j.electacta.2004.12.050
Chen, Z.W., Holmberg, B., Li, W.Z., et al.: Nafion/zeolite nanocomposite membrane by in situ crystallization for a direct methanol fuel cell. Chem. Mater. 18, 5669–5675 (2006). https://doi.org/10.1021/cm060841q
Byun, S.C., Jeong, Y.J., Park, J.W., et al.: Effect of solvent and crystal size on the selectivity of ZSM-5/Nafion composite membranes fabricated by solution-casting method. Solid State Ion. 177, 3233–3243 (2006). https://doi.org/10.1016/j.ssi.2006.09.014
Gribov, E.N., Parkhomchuk, E.V., Krivobokov, I.M., et al.: Supercritical CO2 assisted synthesis of highly selective Nafion-zeolite nanocomposite membranes for direct methanol fuel cells. J. Membr. Sci. 297, 1–4 (2007). https://doi.org/10.1016/j.memsci.2007.03.020
Yildirim, M.H., Curòs, A.R., Motuzas, J., et al.: Nafion®/H-ZSM-5 composite membranes with superior performance for direct methanol fuel cells. J. Membr. Sci. 338, 75–83 (2009). https://doi.org/10.1016/j.memsci.2009.04.009
Yoonoo, C., Dawson, C.P., Roberts, E.P.L., et al.: Nafion®/mordenite composite membranes for improved direct methanol fuel cell performance. J. Membr. Sci. 369, 367–374 (2011). https://doi.org/10.1016/j.memsci.2010.12.030
Zhang, Z.H., Désilets, F., Felice, V., et al.: On the proton conductivity of Nafion-Faujasite composite membranes for low temperature direct methanol fuel cells. J. Power Sources 196, 9176–9187 (2011). https://doi.org/10.1016/j.jpowsour.2011.07.009
Li, X., Roberts, E.P.L., Holmes, S.M., et al.: Functionalized zeolite A-Nafion composite membranes for direct methanol fuel cells. Solid State Ion. 178, 1248–1255 (2007). https://doi.org/10.1016/j.ssi.2007.06.012
Jung, D.H., Cho, S.Y., Peck, D.H., et al.: Preparation and performance of a Nafion®/montmorillonite nanocomposite membrane for direct methanol fuel cell. J. Power Sources 118, 205–211 (2003). https://doi.org/10.1016/S0378-7753(03)00095-8
Song, M.K., Park, S.B., Kim, Y.T., et al.: Nanocomposite polymer membrane based on cation exchange polymer and nano-dispersed clay sheets. Mol. Cryst. Liq. Cryst. 407, 15–23 (2003). https://doi.org/10.1080/744819008
Thomassin, J.M., Pagnoulle, C., Bizzari, D., et al.: Nafion-layered silicate nanocomposite membrane for fuel cell application. E-Polymers 4, 1–13 (2004). https://doi.org/10.1515/epoly.2004.4.1.182
Thomassin, J.M., Pagnoulle, C., Caldarella, G., et al.: Impact of acid containing montmorillonite on the properties of Nafion® membranes. Polymer 46, 11389–11395 (2005). https://doi.org/10.1016/j.polymer.2005.10.018
Thomassin, J., Pagnoulle, C., Bizzari, D., et al.: Improvement of the barrier properties of Nafion® by fluoro-modified montmorillonite. Solid State Ion. 177, 1137–1144 (2006). https://doi.org/10.1016/j.ssi.2006.04.023
Rhee, C.H., Kim, H.K., Chang, H., et al.: Nafion/sulfonated montmorillonite composite: a new concept electrolyte membrane for direct methanol fuel cells. Chem. Mater. 17, 1691–1697 (2005). https://doi.org/10.1021/cm048058q
Lin, Y.F., Yen, C.Y., Hung, C.H., et al.: A novel composite membranes based on sulfonated montmorillonite modified Nafion® for DMFCs. J. Power Sources 168, 162–166 (2007). https://doi.org/10.1016/j.jpowsour.2007.02.079
Kim, T.K., Kang, M., Choi, Y.S., et al.: Preparation of Nafion-sulfonated clay nanocomposite membrane for direct menthol fuel cells via a film coating process. J. Power Sources 165, 1–8 (2007). https://doi.org/10.1016/j.jpowsour.2006.11.055
Lee, W., Kim, H., Kim, T.K., et al.: Nafion based organic/inorganic composite membrane for air-breathing direct methanol fuel cells. J. Membr. Sci. 292, 29–34 (2007). https://doi.org/10.1016/j.memsci.2006.12.051
Lin, Y.F., Yen, C.Y., Ma, C.C.M., et al.: Preparation and properties of high performance nanocomposite proton exchange membrane for fuel cell. J. Power Sources 165, 692–700 (2007). https://doi.org/10.1016/j.jpowsour.2007.01.011
Hasani-Sadrabadi, M.M., Dashtimoghadam, E., Majedi, F.S., et al.: Nafion®/bio-functionalized montmorillonite nanohybrids as novel polyelectrolyte membranes for direct methanol fuel cells. J. Power Sources 190, 318–321 (2009). https://doi.org/10.1016/j.jpowsour.2009.01.043
Hudiono, Y., Choi, S., Shu, S., et al.: Porous layered oxide/Nafion® nanocomposite membranes for direct methanol fuel cell applications. Microporous Mesoporous Mater. 118, 427–434 (2009). https://doi.org/10.1016/j.micromeso.2008.09.017
Meenakshi, S., Sahu, A.K., Bhat, S.D., et al.: Mesostructured-aluminosilicate-Nafion hybrid membranes for direct methanol fuel cells. Electrochim. Acta 89, 35–44 (2013). https://doi.org/10.1016/j.electacta.2012.11.003
Park, Y.S., Yamazaki, Y.: Low methanol permeable and high proton-conducting Nafion/calcium phosphate composite membrane for DMFC. Solid State Ion. 176, 1079–1089 (2005). https://doi.org/10.1016/j.ssi.2004.12.012
Sahu, A.K., Bhat, S.D., Pitchumani, S., et al.: Novel organic-inorganic composite polymer-electrolyte membranes for DMFCs. J. Membr. Sci. 345, 305–314 (2009). https://doi.org/10.1016/j.memsci.2009.09.016
Yang, C., Srinivasan, S., Aricò, A.S., et al.: Composite Nafion/zirconium phosphate membranes for direct methanol fuel cell operation at high temperature. Electrochem. Solid-State Lett. 4, A31 (2001). https://doi.org/10.1149/1.1353157
Bauer, F., Willert-Porada, M.: Microstructural characterization of Zr-phosphate-Nafion® membranes for direct methanol fuel cell (DMFC) applications. J. Membr. Sci. 233, 141–149 (2004). https://doi.org/10.1016/j.memsci.2004.01.010
Bauer, F., Willert-Porada, M.: Characterisation of zirconium and titanium phosphates and direct methanol fuel cell (DMFC) performance of functionally graded Nafion(R) composite membranes prepared out of them. J. Power Sources 145, 101–107 (2005). https://doi.org/10.1016/j.jpowsour.2005.01.063
Hou, H.Y., Sun, G.Q., Wu, Z.M., et al.: Zirconium phosphate/Nafion115 composite membrane for high-concentration DMFC. Int. J. Hydrog. Energy 33, 3402–3409 (2008). https://doi.org/10.1016/j.ijhydene.2008.03.060
Casciola, M., Bagnasco, G., Donnadio, A., et al.: Conductivity and methanol permeability of Nafion-zirconium phosphate composite membranes containing high aspect ratio filler particles. Fuel Cells 9, 394–400 (2009). https://doi.org/10.1002/fuce.200800135
Arbizzani, C., Donnadio, A., Pica, M., et al.: Methanol permeability and performance of Nafion-zirconium phosphate composite membranes in active and passive direct methanol fuel cells. J. Power Sources 195, 7751–7756 (2010). https://doi.org/10.1016/j.jpowsour.2009.07.034
Kim, Y.S., Cho, H.S., Song, M.K., et al.: Characterization of Nafion®/zirconium sulphophenyl phosphate nanocomposite membrane for direct methanol fuel cells. J. Nanosci. Nanotechnol. 8, 4640–4643 (2008). https://doi.org/10.1166/jnn.2008.ic63
Staiti, P., Aricò, A.S., Baglio, V., et al.: Hybrid Nafion-silica membranes doped with heteropolyacids for application in direct methanol fuel cells. Solid State Ion. 145, 101–107 (2001). https://doi.org/10.1016/S0167-2738(01)00919-5
Xu, W.L., Lu, T.H., Liu, C.P., et al.: Low methanol permeable composite Nafion/silica/PWA membranes for low temperature direct methanol fuel cells. Electrochim. Acta 50, 3280–3285 (2005). https://doi.org/10.1016/j.electacta.2004.12.014
Kim, Y.C., Jeong, J.Y., Hwang, J.Y., et al.: Incorporation of heteropoly acid, tungstophosphoric acid within MCM-41 via impregnation and direct synthesis methods for the fabrication of composite membrane of DMFC. J. Membr. Sci. 325, 252–261 (2008). https://doi.org/10.1016/j.memsci.2008.07.039
Thiam, H.S., Daud, W.R.W., Kamarudin, S.K., et al.: Nafion/Pd-SiO2 nanofiber composite membranes for direct methanol fuel cell applications. Int. J. Hydrog. Energy 38, 9474–9483 (2013). https://doi.org/10.1016/j.ijhydene.2012.11.141
Dimitrova, P.: Modified Nafion®-based membranes for use in direct methanol fuel cells. Solid State Ion. 150, 115–122 (2002). https://doi.org/10.1016/s0167-2738(02)00267-9
Antonucci, P.L., Aricò, A.S., Cretı̀, P., et al.: Investigation of a direct methanol fuel cell based on a composite Nafion®-silica electrolyte for high temperature operation. Solid State Ion. 125, 431–437 (1999). https://doi.org/10.1016/S0167-2738(99)00206-4
Adjemian, K.T., Lee, S.J., Srinivasan, S., et al.: Silicon oxide nafion composite membranes for proton-exchange membrane fuel cell operation at 80–140 °C. J. Electrochem. Soc. 149, A256 (2002). https://doi.org/10.1149/1.1445431
Jiang, R.C., Kunz, H.R., Fenton, J.M.: Composite silica/Nafion® membranes prepared by tetraethylorthosilicate sol–gel reaction and solution casting for direct methanol fuel cells. J. Membr. Sci. 272, 116–124 (2006). https://doi.org/10.1016/j.memsci.2005.07.026
Lin, Y.H., Li, H.D., Liu, C.P., et al.: Surface-modified Nafion membranes with mesoporous SiO2 layers via a facile dip-coating approach for direct methanol fuel cells. J. Power Sources 185, 904–908 (2008). https://doi.org/10.1016/j.jpowsour.2008.08.067
Xiao, H.P., Liu, S.H.: Zirconium phosphate (ZrP)-based functional materials: synthesis, properties and applications. Mater. Des. 155, 19–35 (2018). https://doi.org/10.1016/j.matdes.2018.05.041
Vaivars, G., Maxakato, N., Mokrani, T., et al.: Zirconium phosphate based inorganic direct methanol fuel cell. Mater. Sci. 10, 162–165 (2004)
Ozden, A., Ercelik, M., Ozdemir, Y., et al.: Enhancement of direct methanol fuel cell performance through the inclusion of zirconium phosphate. Int. J. Hydrog. Energy 42, 21501–21517 (2017). https://doi.org/10.1016/j.ijhydene.2017.01.188
Sigwadi, R., Dhlamini, M.S., Mokrani, T., et al.: The proton conductivity and mechanical properties of Nafion®/ZrP nanocomposite membrane. Heliyon 5, e02240 (2019). https://doi.org/10.1016/j.heliyon.2019.e02240
Sigwadi, R., Mokrani, T., Msomi, P., et al.: The effect of sulfated zirconia and zirconium phosphate nanocomposite membranes on fuel-cell efficiency. Polymers 14, 263 (2022). https://doi.org/10.3390/polym14020263
Liu, J., Wang, H.T., Cheng, S.A., et al.: Nafion-polyfurfuryl alcohol nanocomposite membranes with low methanol permeation. Chem. Commun. (2004). https://doi.org/10.1039/B315742C
Sungpet, A.: Reduction of alcohol permeation through Nafion® by polypyrrole. J. Membr. Sci. 226, 131–134 (2003). https://doi.org/10.1016/j.memsci.2003.08.015
Shimizu, T., Naruhashi, T., Momma, T., et al.: Preparation and methanol permeability of polyaniline/Nafion composite membrane. Electrochemistry 70, 991–993 (2002). https://doi.org/10.5796/electrochemistry.70.991
Smit, M.A., Ocampo, A.L., Espinosa-Medina, M.A., et al.: A modified Nafion membrane with in situ polymerized polypyrrole for the direct methanol fuel cell. J. Power Sources 124, 59–64 (2003). https://doi.org/10.1016/S0378-7753(03)00730-4
Huang, Q.M., Zhang, Q.L., Huang, H.L., et al.: Methanol permeability and proton conductivity of Nafion membranes modified electrochemically with polyaniline. J. Power Sources 184, 338–343 (2008). https://doi.org/10.1016/j.jpowsour.2008.06.013
Tricoli, V., Carretta, N., Bartolozzi, M.: A comparative investigation of proton and methanol transport in fluorinated ionomeric membranes. J. Electrochem. Soc. 147, 1286 (2000). https://doi.org/10.1149/1.1393351
Kim, D.S., Guiver, M.D., Kim, Y.S.: 2009. Proton exchange membranes for direct methanol fuel cells. In: Liu, H.S., Zhang, J.J. (eds.) Electrocatalysis of Direct Methanol Fuel Cells, pp. 379–416. Wiley, Weinheim (2009). https://doi.org/10.1002/9783527627707.ch10
Karthikeyan, C.S., Nunes, S.P., Prado, L.A.S.A., et al.: Polymer nanocomposite membranes for DMFC application. J. Membr. Sci. 254, 139–146 (2005). https://doi.org/10.1016/j.memsci.2004.12.048
Rambabu, G., Bhat, S.D.: Simultaneous tuning of methanol crossover and ionic conductivity of sPEEK membrane electrolyte by incorporation of PSSA functionalized MWCNTs: a comparative study in DMFCs. Chem. Eng. J. 243, 517–525 (2014). https://doi.org/10.1016/j.cej.2014.01.030
Kreuer, K.D.: On the development of proton conducting polymer membranes for hydrogen and methanol fuel cells. J. Membr. Sci. 185, 29–39 (2001). https://doi.org/10.1016/S0376-7388(00)00632-3
Zhang, H.Q., Li, X.F., Zhao, C.J., et al.: Composite membranes based on highly sulfonated PEEK and PBI: morphology characteristics and performance. J. Membr. Sci. 308, 66–74 (2008). https://doi.org/10.1016/j.memsci.2007.09.045
Wu, H.L., Ma, C.C.M., Kuan, H.C., et al.: Sulfonated poly(ether ether ketone)/poly(vinylpyrrolidone) acid-base polymer blends for direct methanol fuel cell application. J. Polym. Sci. B Polym. Phys. 44, 565–572 (2006). https://doi.org/10.1002/polb.20717
Nunes, S.: Inorganic modification of proton conductive polymer membranes for direct methanol fuel cells. J. Membr. Sci. 203, 215–225 (2002). https://doi.org/10.1016/s0376-7388(02)00009-1
Gaowen, Z., Zhentao, Z.: Organic/inorganic composite membranes for application in DMFC. J. Membr. Sci. 261, 107–113 (2005). https://doi.org/10.1016/j.memsci.2005.03.036
Ponce, M.L., Prado, L., Ruffmann, B., et al.: Reduction of methanol permeability in polyetherketone-heteropolyacid membranes. J. Membr. Sci. 217, 5–15 (2003). https://doi.org/10.1016/S0376-7388(02)00309-5
Meenakshi, S., Bhat, S.D., Sahu, A.K., et al.: Modified sulfonated poly(ether ether ketone) based mixed matrix membranes for direct methanol fuel cells. Fuel Cells 13, 851–861 (2013). https://doi.org/10.1002/fuce.201300022
Peera, S.G., Meenakshi, S., Gopi, K.H., et al.: Impact on the ionic channels of sulfonated poly(ether ether ketone) due to the incorporation of polyphosphazene: a case study in direct methanol fuel cells. RSC Adv. 3, 14048 (2013). https://doi.org/10.1039/c3ra41508b
Guo, Q.H., Pintauro, P.N., Tang, H., et al.: Sulfonated and crosslinked polyphosphazene-based proton-exchange membranes. J. Membr. Sci. 154, 175–181 (1999). https://doi.org/10.1016/S0376-7388(98)00282-8
Saarinen, V., Kallio, T., Paronen, M., et al.: New ETFE-based membrane for direct methanol fuel cell. Electrochim. Acta 50, 3453–3460 (2005). https://doi.org/10.1016/j.electacta.2004.12.022
Chen, J.Y., Cao, J.M., Zhang, R.J., et al.: Modifications on promoting the proton conductivity of polybenzimidazole-based polymer electrolyte membranes in fuel cells. Membranes 11, 826 (2021). https://doi.org/10.3390/membranes11110826
Wainright, J.S., Wang, J.T., Savinell, R.F.: Direct methanol fuel cells using acid doped polybenzimidazole as a polymer electrolyte. In: IECEC 96. Proceedings of the 31st Intersociety Energy Conversion Engineering Conference, pp. 1107–1111. IEEE, Washington (2002). https://doi.org/10.1109/IECEC.1996.553862
Cheng, Y., Zhang, J., Lu, S.F., et al.: Significantly enhanced performance of direct methanol fuel cells at elevated temperatures. J. Power Sources 450, 227620 (2020). https://doi.org/10.1016/j.jpowsour.2019.227620
Hu, M.S., Zhang, B.P., Chen, J.L., et al.: Cross-linked polymer electrolyte membrane based on a highly branched sulfonated polyimide with improved electrochemical properties for fuel cell applications. Int. J. Energy Res. 43, 8753–8764 (2019). https://doi.org/10.1002/er.4879
Choudhury, R.R., Gohil, J.M., Dutta, K.: Poly(vinyl alcohol)-based membranes for fuel cell and water treatment applications: a review on recent advancements. Polym. Adv. Technol. 32, 4175–4203 (2021). https://doi.org/10.1002/pat.5431
Maiti, J., Kakati, N., Lee, S.H., et al.: Where do poly(vinyl alcohol) based membranes stand in relation to Nafion® for direct methanol fuel cell applications? J. Power Sources 216, 48–66 (2012). https://doi.org/10.1016/j.jpowsour.2012.05.057
Wong, C.Y., Wong, W.Y., Loh, K.S., et al.: Development of poly(vinyl alcohol)-based polymers as proton exchange membranes and challenges in fuel cell application: a review. Polym. Rev. 60, 171–202 (2020). https://doi.org/10.1080/15583724.2019.1641514
Yuan, Q., Liu, P., Baker, G.L.: Sulfonated polyimide and PVDF based blend proton exchange membranes for fuel cell applications. J. Mater. Chem. A 3, 3847–3853 (2015). https://doi.org/10.1039/c4ta04910a
Subramanian, M.S., Sasikumar, G.: Sulfonated polyether sulfone-poly(vinylidene fluoride) blend membrane for DMFC applications. J. Appl. Polym. Sci. 117, 801–808 (2010). https://doi.org/10.1002/app.31087
Xue, S., Yin, G.P.: Proton exchange membranes based on poly(vinylidene fluoride) and sulfonated poly(ether ether ketone). Polymer 47, 5044–5049 (2006). https://doi.org/10.1016/j.polymer.2006.03.086
Wang, J.H., Li, N.W., Cui, Z.M., et al.: Blends based on sulfonated poly[bis(benzimidazobenzisoquinolinones)] and poly(vinylidene fluoride) for polymer electrolyte membrane fuel cell. J. Membr. Sci. 341, 155–162 (2009). https://doi.org/10.1016/j.memsci.2009.06.002
Pal, S., Mondal, R., Chatterjee, U.: Sulfonated polyvinylidene fluoride and functional copolymer based blend proton exchange membrane for fuel cell application and studies on methanol crossover. Renew. Energy 170, 974–984 (2021). https://doi.org/10.1016/j.renene.2021.02.046
Kotowicz, J., Węcel, D., Kwilinski, A., et al.: Efficiency of the power-to-gas-to-liquid-to-power system based on green methanol. Appl. Energy 314, 118933 (2022). https://doi.org/10.1016/j.apenergy.2022.118933
Tao, Z.W., Wang, C.Y., Zhao, X.Y., et al.: Progress in high-performance anion exchange membranes based on the design of stable cations for alkaline fuel cells. Adv. Mater. Technol. 6, 2001220 (2021). https://doi.org/10.1002/admt.202001220
Li, C.Q., Baek, J.B.: The promise of hydrogen production from alkaline anion exchange membrane electrolyzers. Nano Energy 87, 106162 (2021). https://doi.org/10.1016/j.nanoen.2021.106162
Zelovich, T., Vogt-Maranto, L., Simari, C., et al.: Non-monotonic temperature dependence of hydroxide ion diffusion in anion exchange membranes. Chem. Mater. 34, 2133–2145 (2022). https://doi.org/10.1021/acs.chemmater.1c03594
Yu, E.H., Scott, K.: Direct methanol alkaline fuel cell with catalysed metal mesh anodes. Electrochem. Commun. 6, 361–365 (2004). https://doi.org/10.1016/j.elecom.2004.02.002
Yu, E.H., Scott, K.: Development of direct methanol alkaline fuel cells using anion exchange membranes. J. Power Sources 137, 248–256 (2004). https://doi.org/10.1016/j.jpowsour.2004.06.004
Yu, E.H., Scott, K.: Direct methanol alkaline fuel cells with catalysed anion exchange membrane electrodes. J. Appl. Electrochem. 35, 91–96 (2005). https://doi.org/10.1007/s10800-004-4061-2
Scott, K., Yu, E., Vlachogiannopoulos, G., et al.: Performance of a direct methanol alkaline membrane fuel cell. J. Power Sources 175, 452–457 (2008). https://doi.org/10.1016/j.jpowsour.2007.09.027
Fujiwara, N., Siroma, Z., Yamazaki, S.I., et al.: Direct ethanol fuel cells using an anion exchange membrane. J. Power Sources 185, 621–626 (2008). https://doi.org/10.1016/j.jpowsour.2008.09.024
Bianchini, C., Bambagioni, V., Filippi, J., et al.: Selective oxidation of ethanol to acetic acid in highly efficient polymer electrolyte membrane-direct ethanol fuel cells. Electrochem. Commun. 11, 1077–1080 (2009). https://doi.org/10.1016/j.elecom.2009.03.022
Yanagi, H., Fukuta, K.: Anion exchange membrane and ionomer for alkaline membrane fuel cells (AMFCs). ECS Trans. 16, 257–262 (2008). https://doi.org/10.1149/1.2981860
Matsuoka, K., Iriyama, Y., Abe, T., et al.: Alkaline direct alcohol fuel cells using an anion exchange membrane. J. Power Sources 150, 27–31 (2005). https://doi.org/10.1016/j.jpowsour.2005.02.020
Danks, T.N., Slade, R.C.T., Varcoe, J.R.: Comparison of PVDF- and FEP-based radiation-grafted alkaline anion-exchange membranes for use in low temperature portable DMFCs. J. Mater. Chem. 12, 3371–3373 (2002). https://doi.org/10.1039/B208627A
Danks, T.N., Slade, R.C.T., Varcoe, J.R.: Alkaline anion-exchange radiation-grafted membranes for possible electrochemical application in fuel cells. J. Mater. Chem. 13, 712–721 (2003). https://doi.org/10.1039/B212164F
Chen, N.J., Lee, Y.M.: Anion exchange polyelectrolytes for membranes and ionomers. Prog. Polym. Sci. 113, 101345 (2021). https://doi.org/10.1016/j.progpolymsci.2020.101345
Xue, J.D., Zhang, J.F., Liu, X., et al.: Toward alkaline-stable anion exchange membranes in fuel cells: cycloaliphatic quaternary ammonium-based anion conductors. Electrochem. Energy Rev. 5, 348–400 (2022). https://doi.org/10.1007/s41918-021-00105-7
Kang, J.J., Li, W.Y., Lin, Y., et al.: Synthesis and ionic conductivity of a polysiloxane containing quaternary ammonium groups. Polym. Adv. Technol. 15, 61–64 (2004). https://doi.org/10.1002/pat.434
Yi, F., Yang, X.P., Li, Y.J., et al.: Synthesis and ion conductivity of poly(oxyethylene) methacrylates containing a quaternary ammonium group. Polym. Adv. Technol. 10, 473–475 (1999). https://doi.org/10.1002/(SICI)1099-1581(199907)10:7%3c473:AID-PAT900%3e3.0.CO;2-2
Li, L., Wang, Y.X.: Sulfonated polyethersulfone Cardo membranes for direct methanol fuel cell. J. Membr. Sci. 246, 167–172 (2005). https://doi.org/10.1016/j.memsci.2004.08.015
Fang, J., Shen, P.K.: Quaternized poly(phthalazinon ether sulfone ketone) membrane for anion exchange membrane fuel cells. J. Membr. Sci. 285, 317–322 (2006). https://doi.org/10.1016/j.memsci.2006.08.037
Zakaria, Z., Kamarudin, S.K.: A review of quaternized polyvinyl alcohol as an alternative polymeric membrane in DMFCs and DEFCs. Int. J. Energy Res. 44, 6223–6239 (2020). https://doi.org/10.1002/er.5314
Xing, B., Savadogo, O.: Hydrogen/oxygen polymer electrolyte membrane fuel cells (PEMFCs) based on alkaline-doped polybenzimidazole (PBI). Electrochem. Commun. 2, 697–702 (2000). https://doi.org/10.1016/S1388-2481(00)00107-7
Hou, H.Y., Sun, G.Q., He, R.H., et al.: Alkali doped polybenzimidazole membrane for alkaline direct methanol fuel cell. Int. J. Hydrog. Energy 33, 7172–7176 (2008). https://doi.org/10.1016/j.ijhydene.2008.09.023
Hou, H.Y., Sun, G.Q., He, R.H., et al.: Alkali doped polybenzimidazole membrane for high performance alkaline direct ethanol fuel cell. J. Power Sources 182, 95–99 (2008). https://doi.org/10.1016/j.jpowsour.2008.04.010
Modestov, A.D., Tarasevich, M.R., Leykin, A.Y., et al.: MEA for alkaline direct ethanol fuel cell with alkali doped PBI membrane and non-platinum electrodes. J. Power Sources 188, 502–506 (2009). https://doi.org/10.1016/j.jpowsour.2008.11.118
Liu, G.L., Wang, A.L., Ji, W.X., et al.: In-situ crosslinked, side chain polybenzimidazole-based anion exchange membranes for alkaline direct methanol fuel cells. Chem. Eng. J. 454, 140046 (2023). https://doi.org/10.1016/j.cej.2022.140046
Yang, C.C., Chiu, S.J., Chien, W.C.: Development of alkaline direct methanol fuel cells based on crosslinked PVA polymer membranes. J. Power Sources 162, 21–29 (2006). https://doi.org/10.1016/j.jpowsour.2006.06.065
Yang, C.C., Lee, Y.J., Chiu, S.J., et al.: Preparation of a PVA/HAP composite polymer membrane for a direct ethanol fuel cell (DEFC). J. Appl. Electrochem. 38, 1329–1337 (2008). https://doi.org/10.1007/s10800-008-9563-x
Yang, C.C., Chiu, S.J., Lee, K.T., et al.: Study of poly(vinyl alcohol)/titanium oxide composite polymer membranes and their application on alkaline direct alcohol fuel cell. J. Power Sources 184, 44–51 (2008). https://doi.org/10.1016/j.jpowsour.2008.06.011
Yang, C.C., Chiu, S.J., Chien, W.C., et al.: Quaternized poly(vinyl alcohol)/alumina composite polymer membranes for alkaline direct methanol fuel cells. J. Power Sources 195, 2212–2219 (2010). https://doi.org/10.1016/j.jpowsour.2009.10.091
Ahmad, A.L., Yusuf, N.M., Ooi, B.S.: Preparation and modification of poly (vinyl) alcohol membrane: effect of crosslinking time towards its morphology. Desalination 287, 35–40 (2012). https://doi.org/10.1016/j.desal.2011.12.003
Herranz, D., Escudero-Cid, R., Montiel, M., et al.: Poly (vinyl alcohol) and poly (benzimidazole) blend membranes for high performance alkaline direct ethanol fuel cells. Renew. Energy 127, 883–895 (2018). https://doi.org/10.1016/j.renene.2018.05.020
Bhat, S.D., Sahu, A.K., George, C., et al.: Mordenite-incorporated PVA-PSSA membranes as electrolytes for DMFCs. J. Membr. Sci. 340, 73–83 (2009). https://doi.org/10.1016/j.memsci.2009.05.014
Yang, T.: Preliminary study of SPEEK/PVA blend membranes for DMFC applications. Int. J. Hydrog. Energy 33, 6772–6779 (2008). https://doi.org/10.1016/j.ijhydene.2008.08.022
Kim, D.S., Park, H.B., Rhim, J.W., et al.: Preparation and characterization of crosslinked PVA/SiO2 hybrid membranes containing sulfonic acid groups for direct methanol fuel cell applications. J. Membr. Sci. 240, 37–48 (2004). https://doi.org/10.1016/j.memsci.2004.04.010
Wang, Y.F., Wang, D., Wang, J.L., et al.: Preparation and characterization of a sol-gel derived silica/PVA-Py hybrid anion exchange membranes for alkaline fuel cell application. J. Electroanal. Chem. 873, 114342 (2020). https://doi.org/10.1016/j.jelechem.2020.114342
Higa, M., Mehdizadeh, S., Feng, S.Y., et al.: Cell performance of direct methanol alkaline fuel cell (DMAFC) using anion exchange membranes prepared from PVA-based block copolymer. J. Membr. Sci. 597, 117618 (2020). https://doi.org/10.1016/j.memsci.2019.117618
Gong, L.Y., Yang, Z.Y., Li, K., et al.: Recent development of methanol electrooxidation catalysts for direct methanol fuel cell. J. Energy Chem. 27, 1618–1628 (2018). https://doi.org/10.1016/j.jechem.2018.01.029
Xia, Z.X., Zhang, X.M., Sun, H., et al.: Recent advances in multi-scale design and construction of materials for direct methanol fuel cells. Nano Energy 65, 104048 (2019). https://doi.org/10.1016/j.nanoen.2019.104048
Tiwari, J.N., Tiwari, R.N., Singh, G., et al.: Recent progress in the development of anode and cathode catalysts for direct methanol fuel cells. Nano Energy 2, 553–578 (2013). https://doi.org/10.1016/j.nanoen.2013.06.009
Lamy, C., Léger, J.M., Srinivasan, S.: Direct methanol fuel cells: from a twentieth century electrochemist’s dream to a twenty-first century emerging technology. In: Modern Aspects of Electrochemistry, pp. 53–118. Boston: Kluwer Academic Publishers (2005). https://doi.org/10.1007/0-306-46923-5_3
Antolini, E., Perez, J.: The renaissance of unsupported nanostructured catalysts for low-temperature fuel cells: from the size to the shape of metal nanostructures. J. Mater. Sci. 46, 4435–4457 (2011). https://doi.org/10.1007/s10853-011-5499-3
Mazumder, V., Lee, Y., Sun, S.H.: Recent development of active nanoparticle catalysts for fuel cell reactions. Adv. Funct. Mater. 20, 1224–1231 (2010). https://doi.org/10.1002/adfm.200902293
Maillard, F., Eikerling, M., Cherstiouk, O.V., et al.: Size effects on reactivity of Pt nanoparticles in CO monolayer oxidation: the role of surface mobility. Faraday Disc. 125, 357 (2004). https://doi.org/10.1039/b303911k
de Sá, M.H., Moreira, C.S., Pinto, A.M.F.R., et al.: Recent advances in the development of nanocatalysts for direct methanol fuel cells. Energies 15, 6335 (2022). https://doi.org/10.3390/en15176335
Lyu, F.L., Cao, M.H., Mahsud, A., et al.: Interfacial engineering of noble metals for electrocatalytic methanol and ethanol oxidation. J. Mater. Chem. A 8, 15445–15457 (2020). https://doi.org/10.1039/d0ta03199b
Aric, A.S., Baglio, V., Antonucci, V.: Direct methanol fuel cells: history, status and perspectives. In: Electrocatalysis of Direct Methanol Fuel Cells, pp. 1–78. Weinheim, Germany: Wiley (2009). https://doi.org/10.1002/9783527627707.ch1
Kakati, N., Maiti, J., Lee, S.H., et al.: Anode catalysts for direct methanol fuel cells in acidic media: do we have any alternative for Pt or Pt-Ru? Chem. Rev. 114, 12397–12429 (2014). https://doi.org/10.1021/cr400389f
Liu, H.S., Song, C.J., Zhang, L., et al.: A review of anode catalysis in the direct methanol fuel cell. J. Power Sources 155, 95–110 (2006). https://doi.org/10.1016/j.jpowsour.2006.01.030
Ren, X.F., Lv, Q.Y., Liu, L.F., et al.: Current progress of Pt and Pt-based electrocatalysts used for fuel cells. Sustain. Energy Fuels 4, 15–30 (2020). https://doi.org/10.1039/c9se00460b
Li, M.Y., Zheng, H.J., Han, G.Y., et al.: Facile synthesis of binary PtRu nanoflowers for advanced electrocatalysts toward methanol oxidation. Catal. Commun. 92, 95–99 (2017). https://doi.org/10.1016/j.catcom.2017.01.014
Chen, F.X., Ren, J.K., He, Q., et al.: Facile and one-pot synthesis of uniform PtRu nanoparticles on polydopamine-modified multiwalled carbon nanotubes for direct methanol fuel cell application. J. Colloid Interface Sci. 497, 276–283 (2017). https://doi.org/10.1016/j.jcis.2017.03.026
Muthuswamy, N., de la Fuente, J.L.G., Tran, D.T., et al.: Ru@Pt core-shell nanoparticles for methanol fuel cell catalyst: control and effects of shell composition. Int. J. Hydrog. Energy 38, 16631–16641 (2013). https://doi.org/10.1016/j.ijhydene.2013.02.056
Xie, J., Zhang, Q.H., Gu, L., et al.: Ruthenium-platinum core-shell nanocatalysts with substantially enhanced activity and durability towards methanol oxidation. Nano Energy 21, 247–257 (2016). https://doi.org/10.1016/j.nanoen.2016.01.013
Liu, H.X., Tian, N., Brandon, M.P., et al.: Tetrahexahedral Pt nanocrystal catalysts decorated with Ru adatoms and their enhanced activity in methanol electrooxidation. ACS Catal. 2, 708–715 (2012). https://doi.org/10.1021/cs200686a
Kakati, N., Lee, S.H., Maiti, J., et al.: Ru decorated Pt nanoparticles by a modified polyol process for enhanced catalytic activity for methanol oxidation. Surf. Sci. 606, 1633–1637 (2012). https://doi.org/10.1016/j.susc.2012.07.008
Burstein, G.T., Barnett, C.J., Kucernak, A.R., et al.: Aspects of the anodic oxidation of methanol. Catal. Today 38, 425–437 (1997). https://doi.org/10.1016/S0920-5861(97)00107-7
Aricò, A.S., Antonucci, P.L., Modica, E., et al.: Effect of Pt-Ru alloy composition on high-temperature methanol electro-oxidation. Electrochim. Acta 47, 3723–3732 (2002). https://doi.org/10.1016/S0013-4686(02)00342-0
Gasteiger, H.A., Marković, N., Ross, P.N., Jr., et al.: Temperature-dependent methanol electro-oxidation on well-characterized Pt–Ru alloys. J. Electrochem. Soc. 141, 1795–1803 (1994). https://doi.org/10.1149/1.2055007
Sgroi, M., Zedde, F., Barbera, O., et al.: Cost analysis of direct methanol fuel cell stacks for mass production. Energies 9, 1008 (2016). https://doi.org/10.3390/en9121008
Lu, Q.Q., Huang, J.S., Han, C., et al.: Facile synthesis of composition-tunable PtRh nanosponges for methanol oxidation reaction. Electrochim. Acta 266, 305–311 (2018). https://doi.org/10.1016/j.electacta.2018.02.021
Yan, R.W., Sun, X.Y., Zhang, X.L., et al.: High quality electrocatalyst by Pd–Pt alloys nanoparticles uniformly distributed on polyaniline/carbon nanotubes for effective methanol oxidation. Nanotechnology 31, 135703 (2020). https://doi.org/10.1088/1361-6528/ab5e94
Gruzeł, G., Szmuc, K., Drzymała, E., et al.: Thin layer vs. nanoparticles: effect of SnO2 addition to PtRhNi nanoframes for ethanol oxidation reaction. Int. J. Hydrog. Energy 47, 14823–14835 (2022). https://doi.org/10.1016/j.ijhydene.2022.02.217
Xia, T.Y., Zhao, K., Zhu, Y.Q., et al.: Mixed-dimensional Pt-Ni alloy polyhedral nanochains as bifunctional electrocatalysts for direct methanol fuel cells. Adv. Mater. 35, 2206508 (2023). https://doi.org/10.1002/adma.202206508
Lou, W.H., Ali, A., Shen, P.K.: Recent development of Au arched Pt nanomaterials as promising electrocatalysts for methanol oxidation reaction. Nano Res. 15, 18–37 (2022). https://doi.org/10.1007/s12274-021-3461-5
Zhang, X.L., Yan, R.W., Zhou, W.C., et al.: Pt–Ru bimetallic nanoparticles anchored on carbon nanotubes/polyaniline composites with coral-like structure for enhanced methanol oxidation. J. Alloys Compd. 920, 165990 (2022). https://doi.org/10.1016/j.jallcom.2022.165990
Bhuvanendran, N., Ravichandran, S., Zhang, W.Q., et al.: Highly efficient methanol oxidation on durable PtxIr/MWCNT catalysts for direct methanol fuel cell applications. Int. J. Hydrog. Energy 45, 6447–6460 (2020). https://doi.org/10.1016/j.ijhydene.2019.12.176
Kaur, A., Kaur, G., Singh, P.P., et al.: Supported bimetallic nanoparticles as anode catalysts for direct methanol fuel cells: a review. Int. J. Hydrog. Energy 46, 15820–15849 (2021). https://doi.org/10.1016/j.ijhydene.2021.02.037
Yu, Y.Q., Chen, K.C., Wu, Q., et al.: Recent progress on reduced graphene oxide supported Pt-based catalysts and electrocatalytic oxidation performance of methanol. Int. J. Hydrog. Energy 48, 1785–1812 (2023). https://doi.org/10.1016/j.ijhydene.2022.10.021
Chen, F., Sun, Y.X., Li, H.Y., et al.: Review and development of anode electrocatalyst carriers for direct methanol fuel cell. Energy Technol. 10, 2101086 (2022). https://doi.org/10.1002/ente.202101086
Martínez-Huerta, M.V., Rojas, S., Gómez de la Fuente, J.L., et al.: Effect of Ni addition over PtRu/C based electrocatalysts for fuel cell applications. Appl. Catal. B Environ. 69, 75–84 (2006). https://doi.org/10.1016/j.apcatb.2006.05.020
Pasupathi, S., Tricoli, V.: Effect of third metal on the electrocatalytic activity of PtRu/Vulcan for methanol electro-oxidation. J. Solid State Electrochem. 12, 1093–1100 (2008). https://doi.org/10.1007/s10008-007-0441-y
Huang, T., Liu, J.L., Li, R.S., et al.: A novel route for preparation of PtRuMe (Me = Fe Co, Ni) and their catalytic performance for methanol electrooxidation. Electrochem. Commun. 11, 643–646 (2009). https://doi.org/10.1016/j.elecom.2009.01.008
Neto, A.O., Franco, E.G., Aricó, E., et al.: New electrocatalysts for electro-oxidation of methanol prepared by Bönnemann’s method. Port. Electrochim. Acta 22, 93–101 (2004). https://doi.org/10.4152/pea.200402093
Wang, Z.B., Zuo, P.J., Yin, G.P.: Investigations of compositions and performance of PtRuMo/C ternary catalysts for methanol electrooxidation. Fuel Cells 9, 106–113 (2009). https://doi.org/10.1002/fuce.200800096
Chai, G.S., Yu, J.S.: Highly efficient Pt–Ru–Co–W quaternary anode catalysts for methanol electrooxidation discovered by combinatorial analysis. J. Mater. Chem. 19, 6842–6848 (2009). https://doi.org/10.1039/B823053F
Scofield, M.E., Koenigsmann, C., Wang, L., et al.: Tailoring the composition of ultrathin, ternary alloy PtRuFe nanowires for the methanol oxidation reaction and formic acid oxidation reaction. Energy Environ. Sci. 8, 350–363 (2015). https://doi.org/10.1039/C4EE02162B
Wang, Q.M., Chen, S.G., Lan, H.Y., et al.: Thermally driven interfacial diffusion synthesis of nitrogen-doped carbon confined trimetallic Pt3CoRu composites for the methanol oxidation reaction. J. Mater. Chem. A 7, 18143–18149 (2019). https://doi.org/10.1039/c9ta04412d
Shang, C.S., Guo, Y.X., Wang, E.K.: Ultrathin nanodendrite surrounded PtRuNi nanoframes as efficient catalysts for methanol electrooxidation. J. Mater. Chem. A 7, 2547–2552 (2019). https://doi.org/10.1039/C9TA00191C
Ma, S.Y., Li, H.H., Hu, B.C., et al.: Synthesis of low Pt-based quaternary PtPdRuTe nanotubes with optimized incorporation of Pd for enhanced electrocatalytic activity. J. Am. Chem. Soc. 139, 5890–5895 (2017). https://doi.org/10.1021/jacs.7b01482
Yang, T.Y., Qin, F.J., Zhang, S.P., et al.: Atomically dispersed Ru in Pt3Sn intermetallic alloy as an efficient methanol oxidation electrocatalyst. Chem. Commun. 57, 2164–2167 (2021). https://doi.org/10.1039/d0cc08210d
Sang, Y.Q., Zhang, R.Y., Xu, B., et al.: Ultrafine and highly dispersed PtRu alloy on polyacrylic acid-grafted carbon nanotube@tin oxide core/shell composites for direct methanol fuel cells. ACS Appl. Energy Mater. 5, 4179–4190 (2022). https://doi.org/10.1021/acsaem.1c03617
Maiyalagan, T., Viswanathan, B.: Catalytic activity of platinum/tungsten oxide nanorod electrodes towards electro-oxidation of methanol. J. Power Sources 175, 789–793 (2008). https://doi.org/10.1016/j.jpowsour.2007.09.106
Maiyalagan, T., Khan, F.N.: Electrochemical oxidation of methanol on Pt/V2O5–C composite catalysts. Catal. Commun. 10, 433–436 (2009). https://doi.org/10.1016/j.catcom.2008.10.011
Zhou, C.M., Wang, H.J., Peng, F., et al.: MnO2/CNT supported Pt and PtRu nanocatalysts for direct methanol fuel cells. Langmuir 25, 7711–7717 (2009). https://doi.org/10.1021/la900250w
Amin, R.S., El-Khatib, K.M., Siracusano, S., et al.: Metal oxide promoters for methanol electro-oxidation. Int. J. Hydrog. Energy 39, 9782–9790 (2014). https://doi.org/10.1016/j.ijhydene.2014.04.100
Chen, W.S., Xue, J., Bao, Y.F., et al.: Surface engineering of nano-ceria facet dependent coupling effect on Pt nanocrystals for electro-catalysis of methanol oxidation reaction. Chem. Eng. J. 381, 122752 (2020). https://doi.org/10.1016/j.cej.2019.122752
Chen, L.G., Liang, X., Li, X.T., et al.: Promoting electrocatalytic methanol oxidation of platinum nanoparticles by cerium modification. Nano Energy 73, 104784 (2020). https://doi.org/10.1016/j.nanoen.2020.104784
Baglio, V., Zignani, S.C., Siracusano, S., et al.: Composite anode electrocatalyst for direct methanol fuel cells. Electrocatalysis 4, 235–240 (2013). https://doi.org/10.1007/s12678-013-0139-0
Baglio, V., Sebastián, D., D’Urso, C., et al.: Composite anode electrode based on iridium oxide promoter for direct methanol fuel cells. Electrochim. Acta 128, 304–310 (2014). https://doi.org/10.1016/j.electacta.2013.10.141
Baglio, V., Amin, R.S., El-Khatib, K.M., et al.: IrO2 as a promoter of Pt–Ru for methanol electro-oxidation. Phys. Chem. Chem. Phys. 16, 10414–10418 (2014). https://doi.org/10.1039/C4CP00466C
Hunt, S.T., Milina, M., Alba-Rubio, A.C., et al.: Self-assembly of noble metal monolayers on transition metal carbide nanoparticle catalysts. Science 352, 974–978 (2016). https://doi.org/10.1126/science.aad8471
Chang, J.F., Feng, L.G., Jiang, K., et al.: Pt-CoP/C as an alternative PtRu/C catalyst for direct methanol fuel cells. J. Mater. Chem. A 4, 18607–18613 (2016). https://doi.org/10.1039/C6TA07896F
Liu, H., Yang, D.W., Bao, Y.F., et al.: One-step efficiently coupling ultrafine Pt-Ni2P nanoparticles as robust catalysts for methanol and ethanol electro-oxidation in fuel cells reaction. J. Power Sources 434, 226754 (2019). https://doi.org/10.1016/j.jpowsour.2019.226754
Long, G.F., Li, X.H., Wan, K., et al.: Pt/CN-doped electrocatalysts: superior electrocatalytic activity for methanol oxidation reaction and mechanistic insight into interfacial enhancement. Appl. Catal. B Environ. 203, 541–548 (2017). https://doi.org/10.1016/j.apcatb.2016.10.055
Zhong, J.P., Hou, C., Li, L., et al.: A novel strategy for synthesizing Fe, N, and S tridoped graphene-supported Pt nanodendrites toward highly efficient methanol oxidation. J. Catal. 381, 275–284 (2020). https://doi.org/10.1016/j.jcat.2019.11.002
Ren, Y.J., Chen, K.C., Zhang, Y.Y., et al.: N-doped carbon confined CoFe@Pt nanoparticles with robust catalytic performance for the methanol oxidation reaction. J. Mater. Chem. A 10, 13345–13354 (2022). https://doi.org/10.1039/d2ta03023c
Koenigsmann, C., Wong, S.S.: One-dimensional noble metal electrocatalysts: a promising structural paradigm for direct methanolfuelcells. Energy Environ. Sci. 4, 1161–1176 (2011). https://doi.org/10.1039/C0EE00197J
Lu, Y.X., Du, S.F., Steinberger-Wilckens, R.: One-dimensional nanostructured electrocatalysts for polymer electrolyte membrane fuel cells: a review. Appl. Catal. B Environ. 199, 292–314 (2016). https://doi.org/10.1016/j.apcatb.2016.06.022
Akhairi, M.A.F., Kamarudin, S.K.: Catalysts in direct ethanol fuel cell (DEFC): an overview. Int. J. Hydrog. Energy 41, 4214–4228 (2016). https://doi.org/10.1016/j.ijhydene.2015.12.145
Antolini, E.: Catalysts for direct ethanol fuel cells. J. Power Sources 170, 1–12 (2007). https://doi.org/10.1016/j.jpowsour.2007.04.009
Tsiakaras, P.E.: PtM/C (M = Sn, Ru, Pd, W) based anode direct ethanol-PEMFCs: structural characteristics and cell performance. J. Power Sources 171, 107–112 (2007). https://doi.org/10.1016/j.jpowsour.2007.02.005
Wang, K., Du, H.Y., Sriphathoorat, R., et al.: Vertex-type engineering of Pt–Cu–Rh heterogeneous nanocages for highly efficient ethanol electrooxidation. Adv. Mater. 30, 1804074 (2018). https://doi.org/10.1002/adma.201804074
Han, S.H., Liu, H.M., Chen, P., et al.: Porous trimetallic PtRhCu cubic nanoboxes for ethanol electrooxidation. Adv. Energy Mater. 8, 1801326 (2018). https://doi.org/10.1002/aenm.201801326
Song, S.Q., Tsiakaras, P.: Recent progress in direct ethanol proton exchange membrane fuel cells (DE-PEMFCs). Appl. Catal. B Environ. 63, 187–193 (2006). https://doi.org/10.1016/j.apcatb.2005.09.018
Zignani, S.C., Baglio, V., Linares, J.J., et al.: Performance and selectivity of PtxSn/C electro-catalysts for ethanol oxidation prepared by reduction with different formic acid concentrations. Electrochim. Acta 70, 255–265 (2012). https://doi.org/10.1016/j.electacta.2012.03.055
Antolini, E., Gonzalez, E.R.: A simple model to assess the contribution of alloyed and non-alloyed platinum and tin to the ethanol oxidation reaction on Pt-Sn/C catalysts: application to direct ethanol fuel cell performance. Electrochim. Acta 55, 6485–6490 (2010). https://doi.org/10.1016/j.electacta.2010.06.035
Chu, Y.Y., Zhang, N., Yang, J.J., et al.: Designed synthesis of thin CeO2 nanowires-supported Pt electrocatalysts with pore-interconnected structure and its high catalytic activity for methanol oxidation. J. Mater. Sci. 53, 2087–2101 (2018)
Xiao, M.L., Li, S.T., Zhao, X., et al.: Enhanced catalytic performance of composition-tunable PtCu nanowire networks for methanol electrooxidation. ChemCatChem 6, 2825–2831 (2014). https://doi.org/10.1002/cctc.201402186
Xiao, M.L., Feng, L.G., Zhu, J.B., et al.: Rapid synthesis of a PtRu nano-sponge with different surface compositions and performance evaluation for methanol electrooxidation. Nanoscale 7, 9467–9471 (2015). https://doi.org/10.1039/C5NR00639B
Chen, X.T., Wang, H., Wang, Y., et al.: Synthesis and electrocatalytic performance of multi-component nanoporous PtRuCuW alloy for direct methanol fuel cells. Catalysts 5, 1003–1015 (2015). https://doi.org/10.3390/catal5031003
Lei, F.L., Li, Z.S., Zhang, L., et al.: Facile synthesis of Pt–Cu (Ni, Co)/GNs-CD and their enhanced electro-catalytic activity for methanol oxidation. J. Electrochem. Soc. 163, F913–F918 (2016). https://doi.org/10.1149/2.1211608jes
Wang, Z.L., Fan, H.S., Liang, H.X., et al.: Microfluidic synthesis and characterization of FePtSn/C catalysts with enhanced electro-catalytic performance for direct methanol fuel cells. Electrochim. Acta 230, 245–254 (2017). https://doi.org/10.1016/j.electacta.2017.01.159
Zhao, S.L., Yin, H.J., Du, L., et al.: Three dimensional N-doped graphene/PtRu nanoparticle hybrids as high performance anode for direct methanol fuel cells. J. Mater. Chem. A 2, 3719–3724 (2014). https://doi.org/10.1039/C3TA14809B
Lv, Q., Xiao, Y., Yin, M., et al.: Reconstructed PtFe alloy nanoparticles with bulk-surface differential structure for methanol oxidation. Electrochim. Acta 139, 61–68 (2014). https://doi.org/10.1016/j.electacta.2014.06.135
Bu, L.Z., Feng, Y.G., Yao, J.L., et al.: Facet and dimensionality control of Pt nanostructures for efficient oxygen reduction and methanol oxidation electrocatalysts. Nano Res. 9, 2811–2821 (2016)
Li, H.H., Fu, Q.Q., Xu, L., et al.: Highly crystalline PtCu nanotubes with three dimensional molecular accessible and restructured surface for efficient catalysis. Energy Environ. Sci. 10, 1751–1756 (2017). https://doi.org/10.1039/C7EE00573C
Huang, L., Zhang, X.P., Wang, Q.Q., et al.: Shape-control of Pt-Ru nanocrystals: tuning surface structure for enhanced electrocatalytic methanol oxidation. J. Am. Chem. Soc. 140, 1142–1147 (2018). https://doi.org/10.1021/jacs.7b12353
Du, S.F., Lu, Y.X., Steinberger-Wilckens, R.: PtPd nanowire arrays supported on reduced graphene oxide as advanced electrocatalysts for methanol oxidation. Carbon 79, 346–353 (2014). https://doi.org/10.1016/j.carbon.2014.07.076
Bu, L.Z., Guo, S.J., Zhang, X., et al.: Surface engineering of hierarchical platinum-cobalt nanowires for efficient electrocatalysis. Nat. Commun. 7, 1–10 (2016). https://doi.org/10.1038/ncomms11850
Cao, X., Wang, N., Han, Y., et al.: PtAg bimetallic nanowires: facile synthesis and their use as excellent electrocatalysts toward low-cost fuel cells. Nano Energy 12, 105–114 (2015). https://doi.org/10.1016/j.nanoen.2014.12.020
Zhao, Y.G., Liu, J.J., Liu, C.G., et al.: Amorphous CuPt alloy nanotubes induced by Na2S2O3 as efficient catalysts for the methanol oxidation reaction. ACS Catal. 6, 4127–4134 (2016). https://doi.org/10.1021/acscatal.6b00540
Zhang, W.Y., Yang, Y., Huang, B.L., et al.: Ultrathin PtNiM (M = Rh, Os, and Ir) nanowires as efficient fuel oxidation electrocatalytic materials. Adv. Mater. 31, 1805833 (2019). https://doi.org/10.1002/adma.201805833
Sriphathoorat, R., Wang, K., Luo, S.P., et al.: Well-defined PtNiCo core-shell nanodendrites with enhanced catalytic performance for methanol oxidation. J. Mater. Chem. A 4, 18015–18021 (2016). https://doi.org/10.1039/C6TA07370K
Zhang, N., Zhu, Y.M., Shao, Q., et al.: Ternary PtNi/PtxPb/Pt core/multishell nanowires as efficient and stable electrocatalysts for fuel cell reactions. J. Mater. Chem. A 5, 18977–18983 (2017). https://doi.org/10.1039/C7TA05130A
Zheng, J., Cullen, D.A., Forest, R.V., et al.: Platinum-ruthenium nanotubes and platinum-ruthenium coated copper nanowires as efficient catalysts for electro-oxidation of methanol. ACS Catal. 5, 1468–1474 (2015). https://doi.org/10.1021/cs501449y
Chang, R., Zheng, L.J., Wang, C.W., et al.: Synthesis of hierarchical platinum–palladium–copper nanodendrites for efficient methanol oxidation. Appl. Catal. B Environ. 211, 205–211 (2017). https://doi.org/10.1016/j.apcatb.2017.04.040
Tao, L., Shi, Y.L., Huang, Y.C., et al.: Interface engineering of Pt and CeO2 nanorods with unique interaction for methanol oxidation. Nano Energy 53, 604–612 (2018). https://doi.org/10.1016/j.nanoen.2018.09.013
Sebastián, D., Stassi, A., Siracusano, S., et al.: Influence of metal oxide additives on the activity and stability of PtRu/C for methanol electro-oxidation. J. Electrochem. Soc. 162, F713–F717 (2015). https://doi.org/10.1149/2.0531507jes
Li, H.D., Pan, Y., Zhang, D., et al.: Surface oxygen-mediated ultrathin PtRuM (Ni, Fe, and Co) nanowires boosting methanol oxidation reaction. J. Mater. Chem. A 8, 2323–2330 (2020). https://doi.org/10.1039/c9ta11745h
Xue, S.F., Deng, W.T., Yang, F., et al.: Hexapod PtRuCu nanocrystalline alloy for highly efficient and stable methanol oxidation. ACS Catal. 8, 7578–7584 (2018). https://doi.org/10.1021/acscatal.8b00366
Yang, P.P., Yuan, X.L., Hu, H.C., et al.: Solvothermal synthesis of alloyed PtNi colloidal nanocrystal clusters (CNCs) with enhanced catalytic activity for methanol oxidation. Adv. Funct. Mater. 28, 1704774 (2018). https://doi.org/10.1002/adfm.201704774
Liu, H.P., Liu, K., Zhong, P., et al.: Ultrathin Pt–Ag alloy nanotubes with regular nanopores for enhanced electrocatalytic activity. Chem. Mater. 30, 7744–7751 (2018). https://doi.org/10.1021/acs.chemmater.8b03085
Huang, L., Zhang, X.P., Han, Y.J., et al.: High-index facets bounded platinum-lead concave nanocubes with enhanced electrocatalytic properties. Chem. Mater. 29, 4557–4562 (2017). https://doi.org/10.1021/acs.chemmater.7b01282
Zeng, J., Francia, C., Gerbaldi, C., et al.: Hybrid ordered mesoporous carbons doped with tungsten trioxide as supports for Pt electrocatalysts for methanol oxidation reaction. Electrochim. Acta 94, 80–91 (2013). https://doi.org/10.1016/j.electacta.2013.01.139
Sebastián, D., Suelves, I., Pastor, E., et al.: The effect of carbon nanofiber properties as support for PtRu nanoparticles on the electrooxidation of alcohols. Appl. Catal. B Environ. 132(133), 13–21 (2013). https://doi.org/10.1016/j.apcatb.2012.11.018
Alegre, C., Sebastián, D., Gálvez, M., et al.: PtRu nanoparticles deposited by the sulfite complex method on highly porous carbon xerogels: effect of the thermal treatment. Catalysts 3, 744–756 (2013). https://doi.org/10.3390/catal3030744
Shi, H.X., Liao, F., Zhu, W.X., et al.: Effective PtAu nanowire network catalysts with ultralow Pt content for formic acid oxidation and methanol oxidation. Int. J. Hydrog. Energy 45, 16071–16079 (2020). https://doi.org/10.1016/j.ijhydene.2020.04.003
Liu, K., Wang, W., Guo, P.H., et al.: Replicating the defect structures on ultrathin Rh nanowires with Pt to achieve superior electrocatalytic activity toward ethanol oxidation. Adv. Funct. Mater. 29, 1806300 (2019). https://doi.org/10.1002/adfm.201806300
Zhu, Y.M., Bu, L.Z., Shao, Q., et al.: Subnanometer PtRh nanowire with alleviated poisoning effect and enhanced C–C bond cleavage for ethanol oxidation electrocatalysis. ACS Catal. 9, 6607–6612 (2019). https://doi.org/10.1021/acscatal.9b01375
Liu, Y.F., Wei, M.J., Raciti, D., et al.: Electro-oxidation of ethanol using Pt3Sn alloy nanoparticles. ACS Catal. 8, 10931–10937 (2018). https://doi.org/10.1021/acscatal.8b03763
Wang, L., Wu, W., Lei, Z., et al.: High-performance alcohol electrooxidation on Pt3Sn-SnO2 nanocatalysts synthesized through the transformation of Pt-Sn nanoparticles. J. Mater. Chem. A 8, 592–598 (2020). https://doi.org/10.1039/c9ta10886f
Chang, Q.W., Kattel, S., Li, X., et al.: Enhancing C–C bond scission for efficient ethanol oxidation using PtIr nanocube electrocatalysts. ACS Catal. 9, 7618–7625 (2019). https://doi.org/10.1021/acscatal.9b02039
Wala, M., Simka, W.: Effect of anode material on electrochemical oxidation of low molecular weight alcohols: a review. Molecules 26, 2144 (2021). https://doi.org/10.3390/molecules26082144
Spendelow, J.S., Wieckowski, A.: Electrocatalysis of oxygen reduction and small alcohol oxidation in alkaline media. Phys. Chem. Chem. Phys. 9, 2654–2675 (2007). https://doi.org/10.1039/B703315J
European Commission: Critical raw materials. https://ec.europa.eu/growth/sectors/raw-materials/areas-specific-interest/critical-raw-materials_en
Tamaki, T., Yamada, Y., Kuroki, H., et al.: Communication: acid-treated nickel-rich platinum-nickel alloys for oxygen reduction and methanol oxidation reactions in alkaline media. J. Electrochem. Soc. 164, F858–F860 (2017). https://doi.org/10.1149/2.1611707jes
Santos, M.C.L., Ottoni, C.A., de Souza, R.F.B., et al.: Methanol oxidation in alkaline medium using PtIn/C electrocatalysts. Electrocatalysis 7, 445–450 (2016)
Fashedemi, O.O., Ozoemena, K.I.: Enhanced methanol oxidation and oxygen reduction reactions on palladium-decorated FeCo@Fe/C core-shell nanocatalysts in alkaline medium. Phys. Chem. Chem. Phys. 15, 20982–20991 (2013). https://doi.org/10.1039/C3CP52601A
Santasalo-Aarnio, A., Kwon, Y., Ahlberg, E., et al.: Comparison of methanol, ethanol and iso-propanol oxidation on Pt and Pd electrodes in alkaline media studied by HPLC. Electrochem. Commun. 13, 466–469 (2011). https://doi.org/10.1016/j.elecom.2011.02.022
Araujo, R.B., Martín-Yerga, D., dos Santos, E.C., et al.: Elucidating the role of Ni to enhance the methanol oxidation reaction on Pd electrocatalysts. Electrochim. Acta 360, 136954 (2020). https://doi.org/10.1016/j.electacta.2020.136954
Roy Chowdhury, S., Ghosh, S., Bhattachrya, S.K.: Enhanced and synergistic catalysis of one-pot synthesized palladium–nickel alloy nanoparticles for anodic oxidation of methanol in alkali. Electrochim. Acta 250, 124–134 (2017). https://doi.org/10.1016/j.electacta.2017.08.050
Gu, Z.L., Xu, H., Bin, D., et al.: Preparation of PdNi nanospheres with enhanced catalytic performance for methanol electrooxidation in alkaline medium. Colloids Surf. A Physicochem. Eng. Asp. 529, 651–658 (2017). https://doi.org/10.1016/j.colsurfa.2017.06.044
Carvalho, L.L., Colmati, F., Tanaka, A.A.: Nickel–palladium electrocatalysts for methanol, ethanol, and glycerol oxidation reactions. Int. J. Hydrog. Energy 42, 16118–16126 (2017). https://doi.org/10.1016/j.ijhydene.2017.05.124
Liu, J., Wang, J., Kong, F.D., et al.: Facile preparation of three-dimensional porous Pd–Au films and their electrocatalytic activity for methanol oxidation. Catal. Commun. 73, 22–26 (2016). https://doi.org/10.1016/j.catcom.2015.09.033
Shang, H.Y., Xu, H., Wang, C., et al.: General synthesis of Pd-pm (pm = Ga, In, Sn, Pb, Bi) alloy nanosheet assemblies for advanced electrocatalysis. Nanoscale 12, 3411–3417 (2020). https://doi.org/10.1039/c9nr10084a
Sakthinathan, S., Thagavelu, K., Tamizhdurai, P., et al.: Activated graphite supported tunable Au-Pd bimetallic nanoparticle composite electrode for methanol oxidation. J. Nanosci. Nanotechnol. 20, 6376–6384 (2020). https://doi.org/10.1166/jnn.2020.18584
Lee, M.J., Kang, J.S., Kang, Y.S., et al.: Understanding the bifunctional effect for removal of CO poisoning: blend of a platinum nanocatalyst and hydrous ruthenium oxide as a model system. ACS Catal. 6, 2398–2407 (2016). https://doi.org/10.1021/acscatal.5b02580
Mansor, M., Timmiati, S.N., Lim, K.L., et al.: Recent progress of anode catalysts and their support materials for methanol electrooxidation reaction. Int. J. Hydrog. Energy 44, 14744–14769 (2019). https://doi.org/10.1016/j.ijhydene.2019.04.100
Wang, T.J., Li, F.M., Huang, H., et al.: Porous Pd-PdO nanotubes for methanol electrooxidation. Adv. Funct. Mater. 30, 2000534 (2020). https://doi.org/10.1002/adfm.202000534
Qiao, W., Yang, X.D., Li, M., et al.: Hollow Pd/Te nanorods for the effective electrooxidation of methanol. Nanoscale 13, 6884–6889 (2021). https://doi.org/10.1039/d1nr01005k
Yu, Z.P., Xu, J.Y., Amorim, I., et al.: Easy preparation of multifunctional ternary PdNiP/C catalysts toward enhanced small organic molecule electro-oxidation and hydrogen evolution reactions. J. Energy Chem. 58, 256–263 (2021). https://doi.org/10.1016/j.jechem.2020.10.016
Xie, S.Q., Deng, L., Huang, H., et al.: One-pot synthesis of porous Pd-polypyrrole/nitrogen-doped graphene nanocomposite as highly efficient catalyst for electrooxidation of alcohols. J. Colloid Interface Sci. 608, 3130–3140 (2022). https://doi.org/10.1016/j.jcis.2021.11.039
Zhang, M.R., Zhu, J.P., Wan, R., et al.: Synergistic effect of nickel oxyhydroxide and tungsten carbide in electrocatalytic alcohol oxidation. Chem. Mater. 34, 959–969 (2022). https://doi.org/10.1021/acs.chemmater.1c02535
Monyoncho, E.A., Woo, T.K., Baranova, E.A.: Ethanol electrooxidation reaction in alkaline media for direct ethanol fuel cells. In: Banks, C., McIntosh, S. (eds.) Electrochemistry, vol. 15, pp. 1–57. Royal Society of Chemistry, Cambridge (2018). https://doi.org/10.1039/9781788013895-00001
Altarawneh, R.M., Brueckner, T.M., Chen, B.Y., et al.: Product distributions and efficiencies for ethanol oxidation at PtNi octahedra. J. Power Sources 400, 369–376 (2018). https://doi.org/10.1016/j.jpowsour.2018.08.052
Lai, S.C.S., Kleijn, S.E.F., Öztürk, F.T.Z., et al.: Effects of electrolyte pH and composition on the ethanol electro-oxidation reaction. Catal. Today 154, 92–104 (2010). https://doi.org/10.1016/j.cattod.2010.01.060
Rao, V., Hariyanto, Cremers, C., et al.: Investigation of the ethanol electro-oxidation in alkaline membrane electrode assembly by differential electrochemical mass spectrometry. Fuel Cells 7, 417–423 (2007). https://doi.org/10.1002/fuce.200700026
Takahashi, H., Sagihara, M., Taguchi, M.: Electrochemically reduced Pt oxide thin film as a highly active electrocatalyst for direct ethanol alkaline fuel cell. Int. J. Hydrog. Energy 39, 18424–18432 (2014). https://doi.org/10.1016/j.ijhydene.2014.09.038
Lai, S.C.S., Koper, M.T.M.: Ethanol electro-oxidation on platinum in alkaline media. Phys. Chem. Chem. Phys. 11, 10446–10456 (2009). https://doi.org/10.1039/B913170A
Zhou, W.: Pt based anode catalysts for direct ethanol fuel cells. Appl. Catal. B Environ. 46, 273–285 (2003). https://doi.org/10.1016/s0926-3373(03)00218-2
Assumpção, M.H.M.T., Nandenha, J., Buzzo, G.S., et al.: The effect of ethanol concentration on the direct ethanol fuel cell performance and products distribution: a study using a single fuel cell/attenuated total reflectance-Fourier transform infrared spectroscopy. J. Power Sources 253, 392–396 (2014). https://doi.org/10.1016/j.jpowsour.2013.12.088
Kim, I., Han, O.H., Chae, S.A., et al.: Catalytic reactions in direct ethanol fuel cells. Angew. Chem. Int. Ed. 50, 2270–2274 (2011). https://doi.org/10.1002/anie.201005745
Zhou, W.J., Song, S.Q., Li, W.Z., et al.: Direct ethanol fuel cells based on PtSn anodes: the effect of Sn content on the fuel cell performance. J. Power Sources 140, 50–58 (2005). https://doi.org/10.1016/j.jpowsour.2004.08.003
Lamy, C., Rousseau, S., Belgsir, E.M., et al.: Recent progress in the direct ethanol fuel cell: development of new platinum-tin electrocatalysts. Electrochim. Acta 49, 3901–3908 (2004). https://doi.org/10.1016/j.electacta.2004.01.078
Varcoe, J.R., Slade, R.C.T., Yee, E.L.H., et al.: Investigations into the ex situ methanol, ethanol and ethylene glycol permeabilities of alkaline polymer electrolyte membranes. J. Power Sources 173, 194–199 (2007). https://doi.org/10.1016/j.jpowsour.2007.04.068
Bianchini, C., Shen, P.K.: Palladium-based electrocatalysts for alcohol oxidation in half cells and in direct alcohol fuel cells. Chem. Rev. 109, 4183–4206 (2009). https://doi.org/10.1021/cr9000995
Ma, L., He, H., Hsu, A., et al.: PdRu/C catalysts for ethanol oxidation in anion-exchange membrane direct ethanol fuel cells. J. Power Sources 241, 696–702 (2013). https://doi.org/10.1016/j.jpowsour.2013.04.051
Shen, S.Y., Zhao, T.S., Xu, J.B., et al.: Synthesis of PdNi catalysts for the oxidation of ethanol in alkaline direct ethanol fuel cells. J. Power Sources 195, 1001–1006 (2010). https://doi.org/10.1016/j.jpowsour.2009.08.079
Moraes, L.P.R., Matos, B.R., Radtke, C., et al.: Synthesis and performance of palladium-based electrocatalysts in alkaline direct ethanol fuel cell. Int. J. Hydrog. Energy 41, 6457–6468 (2016). https://doi.org/10.1016/j.ijhydene.2016.02.150
Bambagioni, V., Bianchini, C., Marchionni, A., et al.: Pd and Pt-Ru anode electrocatalysts supported on multi-walled carbon nanotubes and their use in passive and active direct alcohol fuel cells with an anion-exchange membrane (alcohol = methanol, ethanol, glycerol). J. Power Sources 190, 241–251 (2009). https://doi.org/10.1016/j.jpowsour.2009.01.044
Shen, S.Y., Zhao, T.S., Wu, Q.X.: Product analysis of the ethanol oxidation reaction on palladium-based catalysts in an anion-exchange membrane fuel cell environment. Int. J. Hydrog. Energy 37, 575–582 (2012). https://doi.org/10.1016/j.ijhydene.2011.09.077
Modibedi, R.M., Mehlo, T., Ozoemena, K.I., et al.: Preparation, characterisation and application of Pd/C nanocatalyst in passive alkaline direct ethanol fuel cells (ADEFC). Int. J. Hydrog. Energy 40, 15605–15612 (2015). https://doi.org/10.1016/j.ijhydene.2015.08.113
Hou, H.Y., Wang, S.L., Jiang, Q., et al.: Durability study of KOH doped polybenzimidazole membrane for air-breathing alkaline direct ethanol fuel cell. J. Power Sources 196, 3244–3248 (2011). https://doi.org/10.1016/j.jpowsour.2010.11.104
An, L., Zhao, T.S., Chen, R., et al.: A novel direct ethanol fuel cell with high power density. J. Power Sources 196, 6219–6222 (2011). https://doi.org/10.1016/j.jpowsour.2011.03.040
An, L., Zhao, T.S., Xu, J.B.: A bi-functional cathode structure for alkaline-acid direct ethanol fuel cells. Int. J. Hydrog. Energy 36, 13089–13095 (2011). https://doi.org/10.1016/j.ijhydene.2011.07.025
An, L., Zhao, T.S., Zeng, L., et al.: Performance of an alkaline direct ethanol fuel cell with hydrogen peroxide as oxidant. Int. J. Hydrog. Energy 39, 2320–2324 (2014). https://doi.org/10.1016/j.ijhydene.2013.11.072
Li, Y.S., Zhao, T.S.: A high-performance integrated electrode for anion-exchange membrane direct ethanol fuel cells. Int. J. Hydrog. Energy 36, 7707–7713 (2011). https://doi.org/10.1016/j.ijhydene.2011.03.090
Ma, L., Hsu, A., Chen, R.R.: Performance of PdRu/C anode catalyst for anion-exchange membrane direct ethanol fuel cell. ECS Trans. 58, 1321–1326 (2013). https://doi.org/10.1149/05801.1321ecst
Xu, J.B., Zhao, T.S., Shen, S.Y., et al.: Stabilization of the palladium electrocatalyst with alloyed gold for ethanol oxidation. Int. J. Hydrog. Energy 35, 6490–6500 (2010). https://doi.org/10.1016/j.ijhydene.2010.04.016
Li, Y.S., Zhao, T.S., Chen, R.: Cathode flooding behaviour in alkaline direct ethanol fuel cells. J. Power Sources 196, 133–139 (2011). https://doi.org/10.1016/j.jpowsour.2010.06.111
Vieira, L.E., Jr., Bendo, T., Nieto, M.I., et al.: Processing of copper based foil hardened with zirconia by non-deformation method. Mat. Res. 20, 835–842 (2017). https://doi.org/10.1590/1980-5373-mr-2016-0574
Neto, A.O., da Silva, S.G., Buzzo, G.S., et al.: Ethanol electrooxidation on PdIr/C electrocatalysts in alkaline media: electrochemical and fuel cell studies. Ionics 21, 487–495 (2015)
Tian, N., Zhou, Z.Y., Yu, N.F., et al.: Direct electrodeposition of tetrahexahedral Pd nanocrystals with high-index facets and high catalytic activity for ethanol electrooxidation. J. Am. Chem. Soc. 132, 7580–7581 (2010). https://doi.org/10.1021/ja102177r
Berretti, Giaccherini, Montegrossi, et al.: In-situ quantification of nanoparticles oxidation: a fixed energy X-ray absorption approach. Catalysts 9, 659 (2019). https://doi.org/10.3390/catal9080659
Montegrossi, G., Giaccherini, A., Berretti, E., et al.: Computational speciation models: a tool for the interpretation of spectroelectrochemistry for catalytic layers under operative conditions. J. Electrochem. Soc. 164, E3690–E3695 (2017). https://doi.org/10.1149/2.0711711jes
Berretti, E., Pagliaro, M.V., Giaccherini, A., et al.: Experimental evidence of palladium dissolution in anodes for alkaline direct ethanol and formate fuel cells. Electrochim. Acta 418, 140351 (2022). https://doi.org/10.1016/j.electacta.2022.140351
Hu, C., Zhou, Y.N., Xiao, M.F., et al.: Precise size and dominant-facet control of ultra-small Pt nanoparticles for efficient ethylene glycol, methanol and ethanol oxidation electrocatalysts. Int. J. Hydrog. Energy 45, 4341–4354 (2020). https://doi.org/10.1016/j.ijhydene.2019.11.176
Makin Adam, A.M., Deng, M., Zhu, A.M., et al.: Facile one-step room temperature synthesis of PdAg nanocatalysts supported on multi-walled carbon nanotubes towards electro-oxidation of methanol and ethanol. Electrochim. Acta 339, 135929 (2020). https://doi.org/10.1016/j.electacta.2020.135929
Luo, L.X., Fu, C.H., Yang, F., et al.: Composition-graded Cu-Pd nanospheres with Ir-doped surfaces on N-doped porous graphene for highly efficient ethanol electro-oxidation in alkaline media. ACS Catal. 10, 1171–1184 (2020). https://doi.org/10.1021/acscatal.9b05292
Yang, M., Lao, X.Z., Sun, J., et al.: Assembly of bimetallic PdAg nanosheets and their enhanced electrocatalytic activity toward ethanol oxidation. Langmuir 36, 11094–11101 (2020). https://doi.org/10.1021/acs.langmuir.0c02102
Yang, X.B., Liang, Z.P., Chen, S., et al.: A phosphorus-doped Ag@Pd catalyst for enhanced C–C bond cleavage during ethanol electrooxidation. Small 16, 2004727 (2020). https://doi.org/10.1002/smll.202004727
Lan, B., Huang, M., Wei, R.L., et al.: Ethanol electrooxidation on rhodium-lead catalysts in alkaline media: high mass activity, long-term durability, and considerable CO2 selectivity. Small 16, 2004380 (2020). https://doi.org/10.1002/smll.202004380
Luo, L.X., Fu, C.H., Yan, X.H., et al.: Promoting effects of Au submonolayer shells on structure-designed Cu-Pd/Ir nanospheres: greatly enhanced activity and durability for alkaline ethanol electro-oxidation. ACS Appl. Mater. Interfaces 12, 25961–25971 (2020). https://doi.org/10.1021/acsami.0c05605
Almeida, C.V.S., Tremiliosi-Filho, G., Eguiluz, K.I.B., et al.: Improved ethanol electro-oxidation at Ni@Pd/C and Ni@PdRh/C core-shell catalysts. J. Catal. 391, 175–189 (2020). https://doi.org/10.1016/j.jcat.2020.08.024
Zhang, A., Chen, Y.Y., Yang, Z.P., et al.: Enhanced electrocatalytic activities toward the ethanol oxidation of nanoporous gold prepared via solid-phase reaction. ACS Appl. Energy Mater. 3, 336–343 (2020). https://doi.org/10.1021/acsaem.9b01588
Mozafari, V., Parsa, J.B.: Promoted electrocatalytic performance of palladium nanoparticles using doped-NiO supporting materials toward ethanol electro-oxidation in alkaline media. Int. J. Hydrog. Energy 45, 28847–28859 (2020). https://doi.org/10.1016/j.ijhydene.2020.07.276
Wu, T., Wang, X., Emrehan Emre, A., et al.: Graphene–nickel nitride hybrids supporting palladium nanoparticles for enhanced ethanol electrooxidation. J. Energy Chem. 55, 48–54 (2021). https://doi.org/10.1016/j.jechem.2020.06.056
Liu, D.Y., Zeng, Q., Liu, H., et al.: Combining the core-shell construction with an alloying effect for high efficiency ethanol electrooxidation. Cell Rep. Phys. Sci. 2, 100357 (2021). https://doi.org/10.1016/j.xcrp.2021.100357
Wang, X.M., Zhang, C.M., Chi, M.Z., et al.: Two-dimensional PdSn/TiO2-GO towards ethanol electrooxidation catalyst with high stability. Int. J. Hydrog. Energy 46, 19129–19139 (2021). https://doi.org/10.1016/j.ijhydene.2021.03.058
Hajnajafi, M., Khorshidi, A., Farsadrooh, M., et al.: Nanoscale engineering of building blocks to synthesize a three-dimensional architecture of Pd aerogel as a robust self-supporting catalyst toward ethanol electrooxidation. Energy Fuels 35, 3396–3406 (2021). https://doi.org/10.1021/acs.energyfuels.0c04213
Jiang, M.H., Hu, Y., Zhang, W.J., et al.: Regulating the alloying degree and electronic structure of Pt–Au nanoparticles for high-efficiency direct C2+ alcohol fuel cells. Chem. Mater. 33, 3767–3778 (2021). https://doi.org/10.1021/acs.chemmater.1c00886
Ali Kamyabi, M., Jadali, S.: Rational design of PdCu nanoparticles supported on a templated Ni foam: the cooperation effect of morphology and composition for electrocatalytic oxidation of ethanol. Int. J. Hydrog. Energy 46, 39387–39403 (2021). https://doi.org/10.1016/j.ijhydene.2021.06.106
Lan, B., Wang, Q.L., Ma, Z.X., et al.: Efficient electrochemical ethanol-to-CO2 conversion at rhodium and bismuth hydroxide interfaces. Appl. Catal. B Environ. 300, 120728 (2022). https://doi.org/10.1016/j.apcatb.2021.120728
Wang, C.Q., Bukhvalov, D., Goh, M.C., et al.: Hierarchical AgAu alloy nanostructures for highly efficient electrocatalytic ethanol oxidation. Chin. J. Catal. 43, 851–861 (2022). https://doi.org/10.1016/S1872-2067(21)63895-0
Li, S.W., Zhao, L.M., Shu, J.H., et al.: Mxene coupled over nitrogen-doped graphene anchoring palladium nanocrystals as an advanced electrocatalyst for the ethanol electrooxidation. J. Colloid Interface Sci. 610, 944–952 (2022). https://doi.org/10.1016/j.jcis.2021.11.142
Chen, Y.L., Zhou, Q., Zheng, J.W.: Synergistic decoration of ultrasmall Pd NPs and conductive poly(3,4-ethylenedioxythiophene) coatings on a hydrazone covalent organic framework for boosting ethanol electrooxidation. ACS Sustain. Chem. Eng. 10, 1961–1971 (2022). https://doi.org/10.1021/acssuschemeng.1c08742
Mohammad Mostashari, S., Amiri Dehkharghani, R., Farsadrooh, M., et al.: Engineering three-dimensional superstructure of Pd aerogel with enhanced performance for ethanol electrooxidation. J. Mol. Liq. 360, 119363 (2022). https://doi.org/10.1016/j.molliq.2022.119363
Tang, X.D., Wang, Y.Y., Wang, C.Y., et al.: Pt–Bi on carbon aerogels as efficient electrocatalysts for ethanol oxidation reaction in alkaline medium. J. Alloys Compd. 938, 168398 (2023). https://doi.org/10.1016/j.jallcom.2022.168398
Ali Kamyabi, M., Jadali, S., Alizadeh, T.: Ethanol electrooxidation on nickel foam arrayed with templated PdSn; from catalyst fabrication to electrooxidation dominance route. ChemElectroChem 10, e202200914 (2023). https://doi.org/10.1002/celc.202200914
Ren, X.M., Zelenay, P., Thomas, S., et al.: Recent advances in direct methanol fuel cells at Los Alamos National Laboratory. J. Power Sources 86, 111–116 (2000). https://doi.org/10.1016/S0378-7753(99)00407-3
Küver, A., Vielstich, W.: Investigation of methanol crossover and single electrode performance during PEMDMFC operation. J. Power Sources 74, 211–218 (1998). https://doi.org/10.1016/S0378-7753(98)00065-2
Monteverde Videla, A.H.A., Sebastián, D., Vasile, N.S., et al.: Performance analysis of Fe–N–C catalyst for DMFC cathodes: effect of water saturation in the cathodic catalyst layer. Int. J. Hydrog. Energy 41, 22605–22618 (2016). https://doi.org/10.1016/j.ijhydene.2016.06.060
Cheng, H., Feng, X.L., Wang, D.L., et al.: Synthesis of highly stable and methanol-tolerant electrocatalyst for oxygen reduction: Co supporting on N-doped-C hybridized TiO2. Electrochim. Acta 180, 564–573 (2015). https://doi.org/10.1016/j.electacta.2015.08.143
Colmenares, L., Guerrini, E., Jusys, Z., et al.: Activity, selectivity, and methanol tolerance of novel carbon-supported Pt and Pt3Me (Me = Ni, Co) cathode catalysts. J. Appl. Electrochem. 37, 1413–1427 (2007)
Hernández-Rodríguez, M.A., Goya, M.C., Arévalo, M.C., et al.: Carbon supported Ag and Ag–Co catalysts tolerant to methanol and ethanol for the oxygen reduction reaction in alkaline media. Int. J. Hydrog. Energy 41, 19789–19798 (2016). https://doi.org/10.1016/j.ijhydene.2016.07.188
Nubla, K., Sandhyarani, N.: Ag nanoparticles anchored Ag2WO4 nanorods: an efficient methanol tolerant and durable Pt free electro-catalyst toward oxygen reduction reaction. Electrochim. Acta 340, 135942 (2020). https://doi.org/10.1016/j.electacta.2020.135942
Wang, Y.N., Duan, C.Y., Li, J.H., et al.: Fabrication of interface-engineered Ni/NiO/rGO nanobush for highly efficient and durable oxygen reduction. Mater. Sci. Semicond. Process. 156, 107259 (2023). https://doi.org/10.1016/j.mssp.2022.107259
Ud Din, M.A., Idrees, M., Jamil, S., et al.: Advances and challenges of methanol-tolerant oxygen reduction reaction electrocatalysts for the direct methanol fuel cell. J. Energy Chem. 77, 499–513 (2023). https://doi.org/10.1016/j.jechem.2022.11.023
Antolini, E., Gonzalez, E.R.: Alkaline direct alcohol fuel cells. J. Power Sources 195, 3431–3450 (2010). https://doi.org/10.1016/j.jpowsour.2009.11.145
Liu, L., Pu, C., Viswanathan, R., et al.: Carbon supported and unsupported Pt–Ru anodes for liquid feed direct methanol fuel cells. Electrochim. Acta 43, 3657–3663 (1998). https://doi.org/10.1016/S0013-4686(98)00123-6
Baglio, V., Stassi, A., Matera, F.V., et al.: Optimization of properties and operating parameters of a passive DMFC mini-stack at ambient temperature. J. Power Sources 180, 797–802 (2008). https://doi.org/10.1016/j.jpowsour.2008.02.078
Vasile, N.S., Monteverde Videla, A.H.A., Specchia, S.: Effects of the current density distribution on a single-cell DMFC by tuning the anode catalyst in layers of gradual loadings: modelling and experimental approach. Chem. Eng. J. 322, 722–741 (2017). https://doi.org/10.1016/j.cej.2017.04.060
Vecchio, C.L., Serov, A., Romero, H., et al.: Commercial platinum group metal-free cathodic electrocatalysts for highly performed direct methanol fuel cell applications. J. Power Sources 437, 226948 (2019). https://doi.org/10.1016/j.jpowsour.2019.226948
Osmieri, L., Escudero-Cid, R., Monteverde Videla, A.H.A., et al.: Performance of a Fe–N–C catalyst for the oxygen reduction reaction in direct methanol fuel cell: cathode formulation optimization and short-term durability. Appl. Catal. B Environ. 201, 253–265 (2017). https://doi.org/10.1016/j.apcatb.2016.08.043
Osmieri, L., Escudero-Cid, R., Monteverde Videla, A.H.A., et al.: Application of a non-noble Fe–N–C catalyst for oxygen reduction reaction in an alkaline direct ethanol fuel cell. Renew. Energy 115, 226–237 (2018). https://doi.org/10.1016/j.renene.2017.08.062
Argyropoulos, P., Scott, K., Taama, W.M.: The effect of operating conditions on the dynamic response of the direct methanol fuel cell. Electrochim. Acta 45, 1983–1998 (2000). https://doi.org/10.1016/S0013-4686(99)00420-X
Aricò, A.S., Cretì, P., Modica, E., et al.: Investigation of direct methanol fuel cells based on unsupported Pt–Ru anode catalysts with different chemical properties. Electrochim. Acta 45, 4319–4328 (2000). https://doi.org/10.1016/S0013-4686(00)00531-4
da Silva Freitas, W., Mecheri, B., Lo Vecchio, C., et al.: Metal-organic-framework-derived electrocatalysts for alkaline polymer electrolyte fuel cells. J. Power Sources 550, 232135 (2022). https://doi.org/10.1016/j.jpowsour.2022.232135
Maya-Cornejo, J., Carrera-Cerritos, R., Sebastián, D., et al.: PtCu catalyst for the electro-oxidation of ethanol in an alkaline direct alcohol fuel cell. Int. J. Hydrog. Energy 42, 27919–27928 (2017). https://doi.org/10.1016/j.ijhydene.2017.07.226
Baglio, V., Stassi, A., Modica, E., et al.: Performance comparison of portable direct methanol fuel cell mini-stacks based on a low-cost fluorine-free polymer electrolyte and Nafion membrane. Electrochim. Acta 55, 6022–6027 (2010). https://doi.org/10.1016/j.electacta.2010.05.059
Casalegno, A., Marchesi, R.: DMFC anode polarization: experimental analysis and model validation. J. Power Sources 175, 372–382 (2008). https://doi.org/10.1016/j.jpowsour.2007.09.003
Pasaogullari, U., Wang, C.Y.: Liquid water transport in gas diffusion layer of polymer electrolyte fuel cells. J. Electrochem. Soc. 151, A399 (2004). https://doi.org/10.1149/1.1646148
Lu, Z.J., Daino, M.M., Rath, C., et al.: Water management studies in PEM fuel cells. Part III. Dynamic breakthrough and intermittent drainage characteristics from GDLs with and without MPLs. Int. J. Hydrog. Energy 35, 4222–4233 (2010). https://doi.org/10.1016/j.ijhydene.2010.01.012
Song, J.M., Uchida, H., Watanabe, M.: Effect of wet-proofing treatment of carbon backing layer in gas diffusion electrodes on the PEFC performance. Electrochemistry 73, 189–193 (2005). https://doi.org/10.5796/electrochemistry.73.189
Mehmood, A., An, M.G., Ha, H.Y.: Physical degradation of cathode catalyst layer: a major contributor to accelerated water flooding in long-term operation of DMFCs. Appl. Energy 129, 346–353 (2014). https://doi.org/10.1016/j.apenergy.2014.05.016
Liu, X., Guo, H., Ma, C.F.: Water flooding and two-phase flow in cathode channels of proton exchange membrane fuel cells. J. Power Sources 156, 267–280 (2006). https://doi.org/10.1016/j.jpowsour.2005.06.027
Zago, M., Casalegno, A., Santoro, C., et al.: Water transport and flooding in DMFC: experimental and modeling analyses. J. Power Sources 217, 381–391 (2012). https://doi.org/10.1016/j.jpowsour.2012.06.022
Casalegno, A., Santoro, C., Rinaldi, F., et al.: Low methanol crossover and high efficiency direct methanol fuel cell: the influence of diffusion layers. J. Power Sources 196, 2669–2675 (2011). https://doi.org/10.1016/j.jpowsour.2010.11.050
Hosseinpour, M., Sahoo, M., Perez-Page, M., et al.: Improving the performance of direct methanol fuel cells by implementing multilayer membranes blended with cellulose nanocrystals. Int. J. Hydrog. Energy 44, 30409–30419 (2019). https://doi.org/10.1016/j.ijhydene.2019.09.194
Shu, Q.Z., Xia, Z.X., Wei, W., et al.: Controllable unzipping of carbon nanotubes as advanced Pt catalyst supports for oxygen reduction. ACS Appl. Energy Mater. 2, 5446–5455 (2019). https://doi.org/10.1021/acsaem.9b00506
Choi, B., Nam, W.H., Chung, D.Y., et al.: Enhanced methanol tolerance of highly Pd rich Pd-Pt cathode electrocatalysts in direct methanol fuel cells. Electrochim. Acta 164, 235–242 (2015). https://doi.org/10.1016/j.electacta.2015.02.203
Lo Vecchio, C., Alegre, C., Sebastián, D., et al.: Investigation of supported Pd-based electrocatalysts for the oxygen reduction reaction: performance, durability and methanol tolerance. Materials 8, 7997–8008 (2015). https://doi.org/10.3390/ma8125438
Rivera-Gavidia, L.M., Luis-Sunga, M., Rodríguez, J.L., et al.: Methanol tolerant Pd-Based carbon supported catalysts as cathode materials for direct methanol fuel cells. Int. J. Hydrog. Energy 45, 20673–20678 (2020). https://doi.org/10.1016/j.ijhydene.2020.01.167
Sanij, F.D., Balakrishnan, P., Leung, P., et al.: Advanced Pd-based nanomaterials for electro-catalytic oxygen reduction in fuel cells: a review. Int. J. Hydrog. Energy 46, 14596–14627 (2021). https://doi.org/10.1016/j.ijhydene.2021.01.185
Aricò, A.S., Baglio, V., Di Blasi, A., et al.: Analysis of the high-temperature methanol oxidation behaviour at carbon-supported Pt–Ru catalysts. J. Electroanal. Chem. 557, 167–176 (2003). https://doi.org/10.1016/S0022-0728(03)00369-3
Aricò, A.S., Baglio, V., Di Blasi, A., et al.: Influence of the acid–base characteristics of inorganic fillers on the high temperature performance of composite membranes in direct methanol fuel cells. Solid State Ion. 161, 251–265 (2003). https://doi.org/10.1016/S0167-2738(03)00283-2
Sebastián, D., Serov, A., Artyushkova, K., et al.: High performance and cost-effective direct methanol fuel cells: Fe–N–C methanol-tolerant oxygen reduction reaction catalysts. Chemsuschem 9, 1986–1995 (2016). https://doi.org/10.1002/cssc.201600583
Lo Vecchio, C., Sebastián, D., Lázaro, M., et al.: Methanol-tolerant M-N-C catalysts for oxygen reduction reactions in acidic media and their application in direct methanol fuel cells. Catalysts 8, 650 (2018). https://doi.org/10.3390/catal8120650
Wang, Y.C., Huang, L., Zhang, P., et al.: Constructing a triple-phase interface in micropores to boost performance of Fe/N/C catalysts for direct methanol fuel cells. ACS Energy Lett. 2, 645–650 (2017). https://doi.org/10.1021/acsenergylett.7b00071
Gurau, V., Bluemle, M.J., De Castro, E.S., et al.: Characterization of transport properties in gas diffusion layers for proton exchange membrane fuel cells. J. Power Sources 165, 793–802 (2007). https://doi.org/10.1016/j.jpowsour.2006.12.068
Satjaritanun, P., Zenyuk, I.V.: Water management strategies for PGM-free catalyst layers for polymer electrolyte fuel cells. Curr. Opin. Electrochem. 25, 100622 (2021). https://doi.org/10.1016/j.coelec.2020.08.004
Shi, Q.R., He, Y.H., Bai, X.W., et al.: Methanol tolerance of atomically dispersed single metal site catalysts: mechanistic understanding and high-performance direct methanol fuel cells. Energy Environ. Sci. 13, 3544–3555 (2020). https://doi.org/10.1039/d0ee01968b
Kosmala, T., Bibent, N., Sougrati, M.T., et al.: Stable, active, and methanol-tolerant PGM-free surfaces in an acidic medium: electron tunneling at play in Pt/FeNC hybrid catalysts for direct methanol fuel cell cathodes. ACS Catal. 10, 7475–7485 (2020). https://doi.org/10.1021/acscatal.0c01288
Meenakshi, S., Nishanth, K.G., Sridhar, P., et al.: Spillover effect induced Pt-TiO2/C as ethanol tolerant oxygen reduction reaction catalyst for direct ethanol fuel cells. Electrochim. Acta 135, 52–59 (2014). https://doi.org/10.1016/j.electacta.2014.04.142
Nishanth, K.G., Sridhar, P., Pitchumani, S.: Enhanced oxygen reduction reaction activity through spillover effect by Pt-Y(OH)3/C catalyst in direct methanol fuel cells. Electrochem. Commun. 13, 1465–1468 (2011). https://doi.org/10.1016/j.elecom.2011.09.021
Piela, P., Eickes, C., Brosha, E., et al.: Ruthenium crossover in direct methanol fuel cell with Pt–Ru black anode. J. Electrochem. Soc. 151, A2053 (2004). https://doi.org/10.1149/1.1814472
Ratso, S., Kruusenberg, I., Käärik, M., et al.: Transition metal-nitrogen co-doped carbide-derived carbon catalysts for oxygen reduction reaction in alkaline direct methanol fuel cell. Appl. Catal. B Environ. 219, 276–286 (2017). https://doi.org/10.1016/j.apcatb.2017.07.036
An, L., Zhao, T.S., Li, Y.S.: Carbon-neutral sustainable energy technology: direct ethanol fuel cells. Renew. Sustain. Energy Rev. 50, 1462–1468 (2015). https://doi.org/10.1016/j.rser.2015.05.074
Wang, Y.H., Wang, F., Fang, Y., et al.: Self-assembled flower-like MnO2 grown on Fe-containing urea-formaldehyde resins based carbon as catalyst for oxygen reduction reaction in alkaline direct methanol fuel cells. Appl. Surf. Sci. 496, 143566 (2019). https://doi.org/10.1016/j.apsusc.2019.143566
Fang, Y., Wang, Y.H., Wang, F., et al.: Fe-Mn bimetallic oxides-catalyzed oxygen reduction reaction in alkaline direct methanol fuel cells. RSC Adv. 8, 8678–8687 (2018). https://doi.org/10.1039/C7RA12610G
Liu, Y., Chen, Y.Z., Li, S., et al.: Improved charge transfer in a Mn2O3@Co1.2Ni1.8O4 hybrid for highly stable alkaline direct methanol fuel cells with good methanol tolerance. ACS Appl. Mater. Interfaces 10, 9485–9494 (2018). https://doi.org/10.1021/acsami.8b00613
Liu, Y., Shu, C.Y., Fang, Y., et al.: Two 3D structured Co–Ni bimetallic oxides as cathode catalysts for high-performance alkaline direct methanol fuel cells. J. Power Sources 361, 160–169 (2017). https://doi.org/10.1016/j.jpowsour.2017.06.062
Xie, X.W., Li, Y., Liu, Z.Q., et al.: Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 458, 746–749 (2009). https://doi.org/10.1038/nature07877
Tan, Q., Qu, T., Shu, C.Y., et al.: High-performance polymer fiber membrane based direct methanol fuel cell system with non-platinum catalysts. ACS Sustain. Chem. Eng. 7, 17145–17153 (2019). https://doi.org/10.1021/acssuschemeng.9b03497
Zhiani, M., Gasteiger, H.A., Piana, M., et al.: Comparative study between platinum supported on carbon and non-noble metal cathode catalyst in alkaline direct ethanol fuel cell (ADEFC). Int. J. Hydrog. Energy 36, 5110–5116 (2011). https://doi.org/10.1016/j.ijhydene.2011.01.079
Gaurava, D., Verma, A., Sharma, D.K., et al.: Development of a direct alcohol alkaline fuel cell stack. Fuel Cells 10, 591–596 (2010). https://doi.org/10.1002/fuce.200900039
Grimmer, I., Zorn, P., Weinberger, S., et al.: Ethanol tolerant precious metal free cathode catalyst for alkaline direct ethanol fuel cells. Electrochim. Acta 228, 325–331 (2017). https://doi.org/10.1016/j.electacta.2017.01.087
Osmieri, L., Escudero-Cid, R., Armandi, M., et al.: Fe–N/C catalysts for oxygen reduction reaction supported on different carbonaceous materials Performance in acidic and alkaline direct alcohol fuel cells. Appl. Catal. B Environ. 205, 637–653 (2017). https://doi.org/10.1016/j.apcatb.2017.01.003
Osmieri, L., Zafferoni, C., Wang, L.Q., et al.: Polypyrrole-derived Fe–Co–N–C catalyst for the oxygen reduction reaction: performance in alkaline hydrogen and ethanol fuel cells. ChemElectroChem 5, 1954–1965 (2018). https://doi.org/10.1002/celc.201800420
Wang, L.Q., Lavacchi, A., Bevilacqua, M., et al.: Energy efficiency of alkaline direct ethanol fuel cells employing nanostructured palladium electrocatalysts. ChemCatChem 7, 2214–2221 (2015). https://doi.org/10.1002/cctc.201500189
Seo, S.H., Lee, C.S.: A study on the overall efficiency of direct methanol fuel cell by methanol crossover current. Appl. Energy 87, 2597–2604 (2010). https://doi.org/10.1016/j.apenergy.2010.01.018
Jiang, R.Z., Rong, C., Chu, D.: Determination of energy efficiency for a direct methanol fuel cell stack by a fuel circulation method. J. Power Sources 126, 119–124 (2004). https://doi.org/10.1016/j.jpowsour.2003.08.022
Chu, D., Jiang, R.Z.: Effect of operating conditions on energy efficiency for a small passive direct methanol fuel cell. Electrochim. Acta 51, 5829–5835 (2006). https://doi.org/10.1016/j.electacta.2006.03.017
Jewett, G., Guo, Z., Faghri, A.: Water and air management systems for a passive direct methanol fuel cell. J. Power Sources 168, 434–446 (2007). https://doi.org/10.1016/j.jpowsour.2007.03.052
Vigier, F., Coutanceau, C., Perrard, A., et al.: Development of anode catalysts for a direct ethanol fuel cell. J. Appl. Electrochem. 34, 439–446 (2004)
Pereira, J.P., Falcão, D.S., Oliveira, V.B., et al.: Performance of a passive direct ethanol fuel cell. J. Power Sources 256, 14–19 (2014). https://doi.org/10.1016/j.jpowsour.2013.12.036
Hou, H.Y., Wang, S.L., Jin, W., et al.: KOH modified Nafion 112 membrane for high performance alkaline direct ethanol fuel cell. Int. J. Hydrog. Energy 36, 5104–5109 (2011). https://doi.org/10.1016/j.ijhydene.2010.12.093
An, L., Zhao, T.S.: An alkaline direct ethanol fuel cell with a cation exchange membrane. Energy Environ. Sci. 4, 2213 (2011). https://doi.org/10.1039/c1ee00002k
Kim, J., Momma, T., Osaka, T.: Cell performance of Pd-Sn catalyst in passive direct methanol alkaline fuel cell using anion exchange membrane. J. Power Sources 189, 999–1002 (2009). https://doi.org/10.1016/j.jpowsour.2008.12.108
Yavari, Z., Noroozifar, M., Parvizi, T.: Performance evaluation of anodic nano-catalyst for direct methanol alkaline fuel cell. Environ. Prog. Sustain. Energy 37, 597–604 (2018). https://doi.org/10.1002/ep.12724
Lue, S.J., Wang, W.T., Mahesh, K.P.O., et al.: Enhanced performance of a direct methanol alkaline fuel cell (DMAFC) using a polyvinyl alcohol/fumed silica/KOH electrolyte. J. Power Sources 195, 7991–7999 (2010). https://doi.org/10.1016/j.jpowsour.2010.06.049
Pan, W.H., Lue, S.J., Chang, C.M., et al.: Alkali doped polyvinyl alcohol/multi-walled carbon nano-tube electrolyte for direct methanol alkaline fuel cell. J. Membr. Sci. 376, 225–232 (2011). https://doi.org/10.1016/j.memsci.2011.04.026
Fortune Business Insights: Direct methanol fuel cell market size, share & COVID-19 impact analysis, by components (bipolar plates, current collector, catalyst, membrane), applications (portable, transportation, stationary), and regional forecast, 2021–2028. https://www.fortunebusinessinsights.com/industry-reports/direct-methanol-fuel-cells-market-100779 (2020)
SFC Energy: Quarterly release. https://www.sfc.com/wp-content/uploads/sites/4/SFC_Q3_22_EN.pdf (2022). Accessed 30 Jan 2023
European Commission: 2030 Climate target plan. https://Climate.Ec.Europa.Eu/Eu-Action/European-Green-Deal/2030-Climate-Target-Plan_en
Ensol Systems: EFOY pro direct methanol fuel cells. https://www.ensolsystems.com/products/efoy-pro-direct-methanol-fuel-cells/
EFOY: EFOY fuel cells. https://www.my-efoy.com/en/efoy-fuell-cells/
Takaguchi, H., Ohashi, M.: 1 kW direct methanol fuel cell system. Fujikura Technical Review, 1–4 (2015). https://www.fujikura.co.jp/eng/rd/gihou/backnumber/pages/__icsFiles/afieldfile/2016/02/16/45e_01_1.pdf
Antig Fuel Cell Innovation: Systems. http://www.antig.com/products/products_systems.htm
FuelCellsWork: China: Yuantong debuts a direct methanol fuel cell logistic vehicle. https://fuelcellsworks.com/news/china-yuantong-debuts-a-direct-methanol-fuel-cell-logistics-vehicle/ (2019)
Bellini, M., Berretti, E., Innocenti, M., et al.: 3D titania nanotube array support for water electrolysis palladium catalysts. Electrochim. Acta 383, 138338 (2021). https://doi.org/10.1016/j.electacta.2021.138338
Capozzoli, L., Caprì, A., Baglio, V., et al.: Ruthenium-loaded titania nanotube arrays as catalysts for the hydrogen evolution reaction in alkaline membrane electrolysis. J. Power Sources 562, 232747 (2023). https://doi.org/10.1016/j.jpowsour.2023.232747
Acknowledgements
AL, FV, HM, JF and EB acknowledge the funding from PRIN 2017 by the Italian Ministry MUR Italy (Grant No. 2017YH9MRK). AL, FV, HM, JF, EB, VB acknowledge the Italian Ministry MISE for the FISR 2019 project AMPERE (FISR2019_01294). CS would like to thank the support from the Italian Ministry of Education, Universities and Research (Ministero dell, Istruzione, dell, Università e della Ricerca – MIUR) through the “Rita Levi Montalcini 2018” fellowship (Grant Number PGR18MAZLI).
Funding
Open access funding provided by ICCOM - FIRENZE within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
Alessandro Lavacchi is an editorial board member and Stefania Specchia is an associate editor for Electrochemical Energy Reviews. They were not involved in the editorial review or the decision to publish this article. All authors declare that there are no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Berretti, E., Osmieri, L., Baglio, V. et al. Direct Alcohol Fuel Cells: A Comparative Review of Acidic and Alkaline Systems. Electrochem. Energy Rev. 6, 30 (2023). https://doi.org/10.1007/s41918-023-00189-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41918-023-00189-3