
Abstract
d-Glucaric acid is a potential biobased platform chemical. Previously mainly Escherichia coli, but also the yeast Saccharomyces cerevisiae, and Pichia pastoris, have been engineered for conversion of d-glucose to d-glucaric acid via myo-inositol. One reason for low yields from the yeast strains is the strong flux towards glycolysis. Thus, to decrease the flux of d-glucose to biomass, and to increase d-glucaric acid yield, the four step d-glucaric acid pathway was introduced into a phosphoglucose isomerase deficient (Pgi1p-deficient) Saccharomyces cerevisiae strain. High d-glucose concentrations are toxic to the Pgi1p-deficient strains, so various feeding strategies and use of polymeric substrates were studied. Uniformly labelled 13C-glucose confirmed conversion of d-glucose to d-glucaric acid. In batch bioreactor cultures with pulsed d-fructose and ethanol provision 1.3 g d-glucaric acid L−1 was produced. The d-glucaric acid titer (0.71 g d-glucaric acid L−1) was lower in nitrogen limited conditions, but the yield, 0.23 g d-glucaric acid [g d-glucose consumed]−1, was among the highest that has so far been reported from yeast. Accumulation of myo-inositol indicated that myo-inositol oxygenase activity was limiting, and that there would be potential to even higher yield. The Pgi1p-deficiency in S. cerevisiae provides an approach that in combination with other reported modifications and bioprocess strategies would promote the development of high yield d-glucaric acid yeast strains.
Similar content being viewed by others
Introduction
d-Glucaric acid (d-saccharic acid) is a di-carboxylic acid that can be used for example to produce furan dicarboxylic acid (van Strien et al. 2020) or various polyamides, and polyesters (Sakuta and Nakamura 2019). Biotechnical conversion of d-glucose to d-glucaric acid (or to the conjugate salt d-glucarate) can provide a selective and less energy intensive alternative to chemical production processes (Zhang et al. 2021).
Moon et al. (2009) were the first to engineer Escherichia coli for production of d-glucaric acid. They introduced activities for myo-inositol-1-phosphate synthase, myo-inositol oxygenase and uronate dehydrogenase into E. coli for conversion of D-glucose via glucose-6-phosphate and 1L-myo-inositol-1-phosphate (1 d-myo-inositol 3-phosphate) to myo-inositol, d-glucuronate and finally to d-glucaric acid, resulting in production of 1.1 g L−1 of d-glucaric acid (Table 1). By introducing a polypeptide scaffold to co-localize the pathway enzymes d-glucaric acid concentration was increased to ~ 2.5 g L−1 (Moon et al. 2010). The myo-inositol oxygenase (MIOX) with its di-iron center and low activity was suggested to be the rate-limiting step (Moon et al. 2010). The MIOX activity was subsequently improved by using an N-terminal fusion of small ubiquitin-related modifier (SUMO) to MIOX, showing 75% increase in myo-inositol to d-glucaric acid conversion (Shiue and Prather 2014). Overexpression of myo-inositol-1-phosphate phosphatase from E. coli was tested for further enhancement of the process and the flux of d-glucose from catabolism towards myo-inositol-1-phosphate was redirected by deletion of the phosphoglucose isomerase (Pgi) and glucose 6-phosphate dehydrogenase (Zwf) encoding genes in E. coli (Shiue et al. 2015). This resulted in an increased yield of d-glucaric acid from d-glucose (yield 0.73 g g−1 with titer of 1.19 g L−1, d-xylose as supplementing carbon source). Another approach used to decrease the flux to glycolysis was altering Pfk activity (Brockman and Prather 2015; Gupta et al. 2017; Hou et al. 2020). The d-glucaric acid pathway has also been used as a demonstration pathway for different synthetic biology approaches including use of MAGE (Raman et al. 2014), and small molecule reporter (Rogers and Church 2016). Dynamic pathway regulation with a quorum sensing based system or myo-inositol biosensor (Doong et al. 2018; Verma et al. 2022), regulation of Pgi translation by a d-fructose dependent control system (Qu et al. 2018), and NAD + regeneration system (Su et al. 2020) have been applied for d-glucaric acid production in E. coli. d-Glucaric acid production has also been demonstrated by in vitro conversion (Lee et al. 2016; Petroll et al. 2020; Su et al. 2019). Without myo-inositol addition, volumetric titers have remained between 1 and 2.5 g L−1, although a recent study reported 5.35 g L−1 for intra plus extracellular titer in E. coli (Su et al. 2020) (reviewed by (Chen et al. 2020)).
Yeast are considered advantageous for organic acid production because of their low pH tolerance and robustness (Abbott et al. 2009). Yeasts have also been engineered for production of d-glucaric acid, although more recently than E. coli (Table 1). In 2016 Gupta et al. first engineered Saccharomyces cerevisiae for production of d-glucaric acid by expressing the d-glucaric acid pathway genes coding for inositol monophosphatase, myo-inositol-1-phosphate synthase, myo-inositol oxygenase either from Mus musculus or from Arabidopsis thaliana and uronate dehydrogenase in a opi1 deletion background (Gupta et al. 2016). The engineered yeast produced a maximum titer of 0.56 g L−1 in batch culture, and 0.98 g L−1 in fed-batch. Liu et al. (2016) introduced the pathway into Pichia pastoris and produced 0.107 g L−1 d-glucaric acid from d-glucose. Since these pioneering studies further pathway engineering approaches by e.g. enzyme and expression optimization, use of scaffolds, or by improving viability, have increased d-glucaric acid titers up to 9.5 g L−1 (Fang et al. 2022, 2023; Li et al. 2021; Zhang et al. 2020). In general, co-feeding of myo-inositol has resulted in higher final d-glucaric acid titers, up to 12.96 g L−1 in S. cerevisiae (Chen et al. 2018; Fang et al. 2022; Guo et al. 2022; Gupta et al. 2016; Li et al. 2021; Marques et al. 2020; Zhao et al. 2021), and 6.61 g L−1 in P. pastoris (Liu et al. 2016). The reported yields on d-glucose using yeast are scarce, and typically below 0.1 g g−1, although recently a yield of 0.216 g g−1 (Guo et al. 2022) was reported (Table 1).
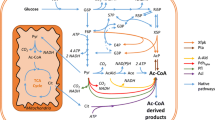
In E. coli deletion of the phosphoglucose isomerase (Pgi) and glucose 6-phosphate dehydrogenase (Zwf) encoding genes resulted in 2.9-fold (Qu et al. 2018), or nearly 18-fold higher yield on d-glucose (Shiue et al. 2015), compared to when the genes were present. The phosphoglucose isomerase encoding gene has not been deleted from S. cerevisiae or P. pastoris strains engineered for d-glucaric acid production. The phosphoglucose isomerase (Pgi1p) -deficient S. cerevisiae strains metabolize d-glucose only poorly, whereas the Pgi-deficient strains of E. coli are able to grow on d-glucose (Vinopal et al. 1975). Even relatively low (< 2 g L−1) d-glucose concentrations inhibit or reduce the growth of S. cerevisiae Pgi1p-deficient strains, possibly because of ATP depletion or glucose-6-phosphate accumulation (Maitra 1971). The accumulation of glucose-6-phosphate, the first metabolite in the d-glucaric acid pathway, could be advantageous for directing d-glucose flux from glycolysis to d-glucaric acid and thus improve the yield of d-glucaric acid on d-glucose. We introduced the d-glucaric acid pathway into a Pgi1p-deficient S. cerevisiae strain (Fig. 1) and studied d-glucaric acid production from monomeric and polymeric d-glucose substrates in shaken flasks and controlled bioreactor conditions with varying nitrogen concentrations. The formation of myo-inositol and d-glucaric acid, and the absence of d-glucuronate was confirmed by using 13C-labelled d-glucose and GC–MS analysis.

Schematic outline for conversion of d-glucose to d-glucaric acid with Pgi1p-deficient S. cerevisiae. In the Pgi1p-deficient strain, d-glucose should be channeled to myo-inositol, while d-fructose and ethanol would allow growth and support the production of biomass. Glucose-6-phosphate can also be channeled to pentose phosphate pathway or to storage carbohydrates
Materials and methods
Strains and strain construction
E. coli strains DH5alpha or TOP10 were used for cloning steps, plasmid propagation and storage, and the Saccharomyces cerevisiae strain FY834 for recombination cloning. S. cerevisiae CEN.PK2-1D (VW-1B; MATα, leu2-3/112 ura3-52 trp1-289 his3∆1 MAL2-8c SUC2) renamed H1346, and the pgi1-deficient strain H2493, described previously (Verho et al. 2002) were the parental strains in which the d-glucaric acid biosynthetic pathway was expressed.
Strain H2493 was cured from tryptophan and/or leucine auxotrophies by transforming it with PCR fragments amplified with primers BCoreMT_LEU2_F and BCoreMT_LEU2_R or BCoreMT_TRP1_F and BCoreMT_TRP1_R (Table 2), using genomic DNA from S. cerevisiae strain S288C as a template, and selecting colonies on plates lacking leucine and/or tryptophan, as appropriate.
The myo-inositol oxygenase encoding gene Miox from Mouse was obtained as a synthetic gene, codon optimized for S. cerevisiae (Gene Art, Germany). S. cerevisiae genomic DNA (from S288C or CEN.PK2-1D) was used to amplify the INO1 (YJL153C) and INM1 (YHR046C) genes (resulting amino acid sequences are the same in both genomes). The uronate dehydrogenase (Udh) gene of Agrobacterium tumefaciens (uro1, GI:223,717,948) (Yoon et al. 2009; Boer et al. 2010) was obtained from the plasmid described by Boer et al. (2010). Miox, INO1 and uro1 were cloned into pRS426 based (B712, (Christianson et al. 1992)) plasmid B5054 (Salusjärvi et al. 2017) that has three promoter-terminator pairs pTEF1 and tADH1, pTPI1 and tCYC1, pPGK1 and tPGK1 by homologous recombination using primers BcoreMT_MIOX_F and BcoreMT_MIOX_R for Miox, BcoreMT_INO1_F and BcoreMT_INO1_R for INO1, and BcoreMT_URO1_F and BcoreMT_URO1_R for uro1 (Table 2), resulting in plasmid B5310. The INM1 gene was expressed either as a single copy genomic integrant or from a multicopy plasmid. INM1 was amplified with primers II_IMP_chr8_BamHI/EcoRI/F and II_IMP_chr8_Bam/R, digested with BamHI and cloned between the PGK1 promoter and terminator in a YEplac195-based multicopy vector B1181 (Toivari et al. 2010a) digested with BglII to create plasmid INM1-B1181. The pPGK1-INM1-tPGK1 cassette was subsequently moved to YEplac181 (B548, (Gietz and Sugino 1988)) by releasing the cassette with HindIII and ligating it to the HindIII site of YEplac181 resulting in plasmid B5154. For genomic integration the INM1 was targeted to GRE3 locus. The 315 bp and and 290 bp regions of GRE3 gene were amplified from the H1346 genomic DNA with primer pairs S.c GRE3 nt-5frw and S.c GRE3 nt310rev and S.c GRE3 nt677frw and S.c GRE3 nt966rev, respectively, where the numbers are relative to nucleotide A in ATG of the GRE3 gene. The BamHI site for cloning the gene expression cassette was included in the 315 bp region. The 315 bp region was ligated into a PvuII and BsiWI linearised plasmid pUG6 (Gueldener et al. 2002), and the plasmid obtained was then cut with EcoRV and SpeI for introducing the 290 bp region. The resulting plasmid, containing the KanMX cassette flanked by the S.cerevisiae GRE3 regions, was named pMLV84. The pMLV84 was digested with BamHI and the pPGK1-INM1-tPGK1 cassette digested with HindIII from INM1-B1181, both fragments were blunt-ended and ligated resulting in INM1-pMLV84. The integration cassette, released with NotI, was transformed into the pgi1-deficient strain cured for TRP and LEU. A cassette without INM1 was transformed to create a control strain.
The INO1-Miox-uro1 B5310 plasmid was introduced to the parental strain H1346 (with intact PGI1) with or without the INM1 plasmid B5154, resulting in strains H4254 and H4355, respectively. The INM1 was integrated to the GRE3 locus of H1346 as described for the pgi1 strains, and the plasmid B5310 was introduced, resulting in strain H5156. The INO1-Miox-uro1 plasmid B5310 was introduced to the pgi1-deficient strain cured with TRP1 and LEU2, with or without INM1 integration, resulting in strains H4350 and H4344, respectively. The B5310 plasmid was also introduced to the pgi1-deficient strain cured for TRP, together with the INM1 plasmid B5154, resulting in strain H4346 or to pgi1-deficient strain with KMX integrated into GRE3, resulting in strain H4351. Corresponding control strains with empty vectors B712 + B548 H4345, and H4349 were also created. The final strains used in the study are listed in Table 3.
Media and culture conditions
Yeast strains were cultured in 20 or 50 mL volume on modified synthetic complete medium (YSC, (Sherman et al. 1983)) without uracil and/or leucine, with 2% (w/v) d-glucose or the d-glucose and d-fructose concentrations indicated in the results, in 100 or 250 mL Erlenmeyer flasks, respectively, at 250 rpm, 30 °C. The pgi1-deficient strains were grown with 0.5 g d-glucose L−1 and 20 g d-fructose L−1 as carbon source for maintenance and biomass generation. The (NH4)2SO4 concentration in YSC was reduced from 5 to 1.5 g L−1 for nitrogen-restricted batch cultures. The flask cultures with pgi1-deficient strains were buffered by CaCO3 (1%, w/v). For slow glucose release, EnBase B (Biosilta, Oulu, Finland) was prepared according to manufacturer’s instructions, except pH was adjusted to pH 5.6 with HCl and enzyme (5 µL to 20 mL culture broth) added after 24 h incubation, because d-glucose (~ 1 g L−1) was present in the prepared medium. Cellulose (α-cellulose, Sigma) was provided as 16.6 g L−1, with 4.5 mL Cellulast 1.5 L added to a volume of 250 mL in bioreactor culture.
For larger scale cultures, yeast were grown in 250 to 500 mL medium (YSC-ura, or YSC-ura-leu) in Multifors bioreactors (max. working volume 500 mL, Infors HT, Switzerland) at pH 5.5, 30 °C, 1 volume air [volume culture]−1 min−1 (vvm) and 500 rpm agitation with 2 marine impellors, as previously described (Toivari et al. 2010a, b). The pH was maintained constant by addition of 2 M NaOH or 1 M H2PO4. Clerol antifoaming agent (Cognis, France, 0.08–0.10 µL−1) was added to prevent foam formation. d-Glucose concentration in the culture supernatant was monitored by HPLC and d-glucose, d-fructose and/or ethanol were added as pulses to keep the d-glucose concentration between 0.5 and 5 g L−1, while providing d-fructose or ethanol as an energy and carbon source for growth.
Biomass was measured as optical density (OD) at 600 nm (OD600) or as dry weight. For dry weight, samples were collected in 2 mL pre-dried, pre-weighed microcentrifuge tubes, washed twice with equal volume distilled water and dried at 105 °C.
The number of metabolically active (vital) cells was determined microscopically by methylene blue (0.25 g L−1 in 0.04 M NaCitrate buffer pH 8.3) staining. For the purpose of clarity the metabolically active cells will be referred to as viable and the inactive as metabolically inactive cells. Both empty and stained cell were counted as metabolically inactive cells.
Chemical analyses
For determination of intracellular d-glucaric acid concentration, cells were collected from 10 mL culture. Cell pellets were washed with 1.8 mL of 0.9% w/v NaCl solution (9 g L−1), and then 1.8 mL deionised water, and frozen at − 20 °C to disrupt membranes. The frozen pellets were freeze-dried using a Christ Alpha 2–4 lyophiliser (Biotech international, Belgium), removing all excess moisture. Intracellular d-glucaric acid was extracted from the lyophilized pellets (6 to 45 mg biomass) in 5 mM H2SO4 (0.5 mL) as described by Nygård et al. (2011) for extraction of d-xylonate. Cell debris was removed by centrifugation and the supernatant analysed by GC–MS. Intracellular concentrations are given as mg per g dry biomass. For a conservative estimate of intracellular concentration, assume that 1 g dry cell weight corresponds to 2 mL cell volume (de Koning and van Dam 1992; Gancedo and Serrano 1989). This estimate is conservative since it does not take into account the volume of intracellular organelles, variation in cell wall thickness, or the contribution of dead cells to the dry biomass.
Concentrations of d-glucose and d-fructose, ethanol, glycerol, and acetate, were analysed by HPLC using a Fast Acid Analysis Column (100 mm × 7.8 mm, BioRad Laboratories, Hercules, CA) linked to an Aminex HPX-87H column (BioRad Labs, USA) with 5 mM H2SO4 as eluent and a flow rate of 0.3 mL min−1. The column was maintained at 55 °C. Peaks were detected using a Waters 410 differential refractometer and a Waters 2487 dual wavelength UV (210 nm) detector. The ability of d-glucaric acid and its dissociated form d-glucarate to form glucaric acid 1,4-lactone or dilactone needs to be considered in the analytics. d-Glucaric acid, glucaric acid 1,4-lactone, d-glucuronate and myo-inositol were quantified with GC–MS. The d-glucaric acid concentration is presented as the sum of d-glucaric acid and glucaric acid 1,4-lactone. Samples (100 µL), with arabitol as internal standard, were evaporated to dryness and silylated by adding 100 µL pyridine, 100 µL chlorotrimethylsilane and 100 µL N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA). Derivatisation was performed at + 70 °C for 60 min. Derivatised samples (1 µL) were subjected to GC–MS analysis (Agilent 6890 Series, USA combined with Agilent, 5973 Network MSD, USA and Combipal injector, Varian Inc., USA). Analytes were injected on split mode (30:1) (200 °C) and separated on a ZB-1HT INFERNO capillary column (30 m × 0.25 mm) with a phase thickness 0.25 µm (Phenomenex, Denmark). Helium (0.9 mL min−1) was used as carrier gas in constant flow mode. The temperature program started at 70 °C with 3 min holding time and then increased 10 °C min−1 up to 320 °C. Mass selective detector (MSD) was operated in electron-impact mode at 70 eV, in the full scan m/z 40–550. The ion source temperature was 250 °C and the interface was 280 °C. Compounds were identified according to corresponding standards and with the Palisade Complete 600 K Mass spectral library (Palisade Mass Spectrometry, USA).
d-Glucaric acid concentrations were also measured as the lactone using the hydroxymate method (Lien 1959) as described by Toivari et al. (2010b). The lactone assay was used for analysing samples from cultures grown with cellulose and although it correlated well with GC–MS results (Fig. 2), the assay would also measure d-gluconic acid, which was probably present in the sample. Thus, lactone measurements should be considered as indicative, but may be over-estimates.

Correlation between d-glucaric acid measured by GC–MS and the lactone measured using the hydroxylamate assay, with background subtracted
Results
d-Glucose conversion to d-glucaric acid with pgi1-deficient S. cerevisiae
The pgi1-deficient S. cerevisiae CEN.PK2-1D strains expressed INO1, Miox and uro1 from a multicopy plasmid and INM1 either as the native genomic copy (H4344), from an additional integrated copy under a constitutive promoter (H4350), or from a multicopy plasmid (H4346) (Table 3). Strains H4349 or H4345 without the glucarate pathway were used as controls. Similar strains, with the d-glucaric acid pathway, but with intact PGI1 gene (H4354, H4355, H4356), were also created (Table 3).
The strains with intact PGI1 gene were grown in flask cultures with 2% (w/v) d-glucose as a sole carbon source. In 48 h at most 35 mg d-glucaric acid L−1 was produced (data not shown).
Although d-glucose inhibits the growth of pgi1-deficient strains at concentrations above 2 g L−1, concentrations up to 4 or 5 g L−1 could be tolerated in cultures with sufficient biomass (OD600 12). With 4 g d-glucose L−1 and 20 g d-fructose L−1, strain H4344, with the d-glucaric acid pathway with endogenous INM1, produced 240 ± 11 mg d-glucaric acid L−1 in 98 h, whereas the control strain H4345 did not produce any d-glucaric acid. In addition, 370 ± 11 mg L−1 of myo-inositol was detected from strain H4344 compared to 100 ± 26 mg L−1 from H4345. All d-glucose and d-fructose were consumed, biomass, ethanol, acetate and glycerol were formed and partly consumed.
The other pgi1-deficient S. cerevisiae strains H4346, with the d-glucaric acid pathway with INM1 on a multicopy plasmid, and the control strain H4349 were cultivated in the presence of 3.4 g L−1 uniformly labelled d-glucose. To decrease biomass formation, lower d-fructose (12 g L−1) and ammonium sulphate concentrations were used with initial high biomass (OD600 13). In 73 h, 280 mg L−1 of labelled d-glucaric acid and 330 mg L−1 labelled myo-inositol was detected. Thus, altogether, 610 mg L−1 of 13C-labelled d-glucose was directed to the d-glucaric acid pathway confirming formation of d-glucaric acid from d-glucose via myo-inositol. The d-glucaric acid pathway intermediate d-glucuronate was not detected. With the control strain H4349 no d-glucaric acid, or d-glucuronate, was detected. With higher d-glucose concentration the pgi1-deficient strains lost viability (data not shown).
Evaluation of different feeding strategies for production of d-glucaric acid in bioreactor cultures
The pgi1-deficient strains were further studied in bioreactor cultures with d-glucose pulses in nitrogen sufficient or restricted conditions. When pgi1-deficient S. cerevisiae strain H4346 expressing the d-glucaric acid pathway was grown in pH regulated batch cultures at pH 5.5, 1.3 g d-glucaric acid L−1 (yield 0.12 ± 0.01 g d-glucaric acid [g d-glucose consumed]−1) were produced (H4346, Figs. 3, 4a). d-Glucaric acid was produced at a rate of approximately 7 mg L−1 h−1. Myo-inositol was detectable in the culture supernatant after 45 h (0.06 g L−1) and accumulated in proportion (10 ± 1%) to d-glucaric acid to a final concentration of 0.15 g L−1. Up to 0.7 g glycerol L−1 and 2.5 g acetate L−1 were also produced, but acetate was consumed when ethanol was not available as a carbon source. Only 4.7 ± 0.1 g biomass L−1 was produced from the 15.9 g d-fructose L−1, 14.7 g d-glucose L−1 and 6.1 g ethanol L−1 which were provided (yield of biomass on substrate ~ 0.17 g [g substrate consumed]−1).

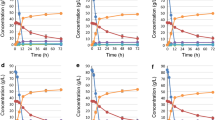
d-Glucaric acid and myo-inositol concentrations (left), d-glucaric acid yield on glucose (middle) and the proportion of viable cells (right) in the culture after 70 h cultivation for two d-glucaric acid producing strains (H4346 and H4350). Cultures were grown in bioreactors at pH 5.5 with pulsed addition of d-glucose (with d-fructose or ethanol for energy) or in fed-batch culture (FB) or a batch culture with cellulose as the source of d-glucose. The nitrogen supply was reduced in one culture receiving pulses of d-glucose. Maximum d-glucaric acid titers were observed after 160, 160, 117, 117 and 191 h for the 5 cultures shown. Yield was not determined for the cellulose batch culture

S. cerevisiae pgi1-deficient strain with a pathway for conversion of d-glucose to d-glucaric acid (pathway genes expressed on multicopy plasmids, H4346), was grown in SC -ura -leu a nitrogen sufficient or b nitrogen-restricted medium at pH 5.5 with d-glucose, d-fructose and ethanol as carbon sources (upper graphs), added initially and as pulses at intervals, as shown. Lower graphs show the concentrations of d-glucaric acid, myo-inositol and biomass produced. Data on product formation by a strain with INM1 integrated (H4350) are also shown (small symbols) for nitrogen-restricted medium. Substrate concentrations and feeding were similar for H4350 and H4346 and are only shown for H4346 on the upper graph. Error bars (biomass) represent ± sem (n = 3) and where not visible were smaller than the symbol
The intracellular concentration of d-glucaric acid was constant at 10 ± 1 mg [g biomass]−1. Intracellular myo-inositol concentrations (6 ± 1 mg [g biomass]−1) were similar to those of d-glucaric acid, and the ratio of myo-inositol to d-glucaric acid in the cytoplasm was much higher than in the supernatant.
Methylene blue staining showed that 63 ± 2% of the cells were metabolically inactive after 70 h (Fig. 3). A high proportion of metabolically inactive cells was also observed for the pgi1-deficient parent strain without the d-glucaric acid pathway in flask cultures (58 ± 2%, after 43 h on SC medium with 12 g d-fructose L−1 and 0.5 g d-glucose L−1 and 92 ± 1% metabolically inactive cells with 12 g d-fructose L−1 and 3.4 g d-glucose L−1), indicating that the high level of metabolically inactive cells was a response to the relatively high concentration of d-glucose in the medium, not to d-glucaric acid production. Some cells adapted to the presence of d-glucose in the medium after ~ 90 h in the bioreactor and the proportion of metabolically inactive cells did not increase much after 70 h.
The pgi1-deficient d-glucaric acid strain H4346 produced 0.71 g d-glucaric acid L−1 after 160 h, with a yield of 0.23 g d-glucaric acid [g d-glucose consumed]−1, in nitrogen-restricted medium (Fig. 4b). There was 0.12 g myo-inositol L−1 also produced. No adaptation to the presence of d-glucose occurred, and this strain produced only 3.7 ± 0.1 g biomass L−1, with 91 ± 0.5% of cells metabolically inactive after 138 h. The concentration of intracellular d-glucaric acid (8 ± 2 mg [g biomass]−1) and myo-inositol (9 ± 4 mg [g biomass]−1) were similar to those observed in the nitrogen sufficient culture. Another pgi1-deficient d-glucaric acid strain with INM1 integrated (H4350) followed a similar trend in d-glucaric acid production but produced less biomass and myo-inositol (Fig. 4b, lower panel).
To lower the d-glucose toxicity by fed-batch cultivation the pgi1-deficient d-glucaric acid strains, H4350 and H4346 were grown with d-glucose and d-fructose in the feed. d-Glucose concentration was maintained at less than 2.2 g L−1 during the first 120 h feeding, with d-fructose concentrations less than 1.5 g L−1 The strains produced 0.32–0.34 g d-glucaric acid L−1 after 117 h, with yield of 0.12 g d-glucaric acid [g d-glucose consumed]−1, but no further production after 117 h. The d-glucose concentration increased after 117 h, reaching concentrations of 3.7–4.5 g L−1 by 160 h (data not shown). Cell viability was measured at 138 h, with 63 ± 4% and 71 ± 4% metabolically inactive cells for strains H4350 and H4346, respectively.
Polymeric substrates for production of d-glucaric acid
To evaluate use of cheap polymeric substrates for production of d-glucaric acid by the pgi1-deficient S. cerevisiae strains a commercial polysaccharide and α-cellulose were used as carbon sources and hydrolytic enzymes used for d-glucose release during d-glucaric acid production.
The d-glucose slowly released from the commercial polysaccharide was rapidly consumed and no d-glucose was detected in the culture media in shake flask cultures. In 100 h 12 g L−1 of d-glucose (as approximated from medium without cells) was released. To decrease production of biomass the amount of nitrogen provided was reduced. d-Fructose provided as a carbon source was consumed during the first 20 h. The pgi1-deficient, d-glucaric acid-pathway-expressing strains H4346 and H4351 produced 0.79 and 0.51 g L−1 d-glucaric acid within 100 h, respectively. Myo-inositol (~ 0.2 g L−1) and biomass were also formed. In bioreactors a maximum ~ 0.65 g d-glucaric acid L−1 was produced from the commercial polysaccharide, and 0.32 (pH 5.5) and 0.35 (pH 7) g d-glucaric acid L−1 was produced from α-cellulose within less than 250 h (Fig. 5a, lactone assay). In the cellulose pH 5.5 culture d-glucose started to accumulate from the beginning of the culture, and in the culture with the commercial polysaccharide after 50 h, whereas with cellulose pH 7 culture d-glucose remained low throughout the culture (Fig. 5b).

d-Glucaric acid production in bioreactor cultures with polymeric substrates. a d-Glucaric acid production. b d-Glucose release and consumption. Cellulose with hydrolytic enzyme in pH 5.5 (open square) or in pH 7 (filled square), commercial polysaccharide with hydrolytic enzyme (Sol glucan pH 7)
Discussion
In S. cerevisiae the major flux of d-glucose is through glycolysis, and only a small fraction, approximately 1–4%, is channelled to the PPP, depending on strain and culture conditions (Blank et al. 2005; Fiaux et al. 2003; Gancedo and Lagunas 1973; Maaheimo et al. 2001; Nidelet et al. 2016). In the pgi1-deficient S. cerevisiae strains, the block to glycolysis leads to accumulation of glucose-6-phosphate (e.g. Ciriacy and Breitenbach 1979; Heux et al. 2008; Maitra 1971). Possibly, this glucose-6-phosphate accumulation could increase the flux of glucose-6-phosphate to myo-inositol and further to d-glucaric acid. Indeed, the yield of d-glucaric acid on d-glucose (0.12 g g−1) in the bioreactor with pulsed d-glucose was four-fold higher in our pgi1-deficient strains compared to the value in a strain with intact Pgi1p reported by Gupta et al. (2016) and was further increased to 0.23 g g−1 (seven fold improvement over Gupta et al. (2016)) by restricting the nitrogen supply. Recently, Guo et al. reported a yield of 0.216 g g−1 (Guo et al. 2022), close to our best yield. However, considering that our strains accumulated inositol at 12–17% of the d-glucaric acid concentration, it is clear that the yield could be even higher if all myo-inositol would be converted to d-glucaric acid. In S. cerevisiae, the PGI1 deletion alone increased the yield, compared to E. coli where the zwf deletion was also needed (Shiue et al. 2015). In yeast, deletion or downregulation of both PGI1 and glucose-6-phosphate dehydrogenase encoding gene ZWF1 has not been reported, but during preparation of this work Zhao et al. (2023) reported that downregulation of ZWF1 in S. cerevisiae improved d-glucaric acid titer by 22.4%.
In volumetric terms our pgi1-deficient strains produced about half of the d-glucaric acid concentration reported by Gupta et al. (2016) in shake flasks (average 0.26 compared to 0.54 g L−1), as confirmed by using 13C-labelled d-glucose. The titer was improved to 1.3 g L−1 in the bioreactor by providing the d-glucose in pulses, which was higher than reported by Gupta et al. (2016), but lower compared to recently reported volumetric titers of up to 9.5 g L−1 (Table 1). Also, the highest yield was observed with restricted nitrogen supply, and in this condition the volumetric concentration was only 0.71 g L−1. Interestingly, our parent strains with intact Pgi1p produced only ~ 30 mg L−1 of d-glucaric acid as measured with GC–MS. This in line with the results of Liu et al. (2016) who found that P. pastoris produced 108 mg L−1 d-glucaric acid on d-glucose, but much lower compared to other studies (Table 1). In our strains the OPI1 gene was intact. We hypothesized that expression of INO1 under a constitutive promoter is comparable to the effect of OPI1 deletion because Opi1p is reported to regulate INO1 at the transcriptional level (Henry et al. 2014; Ye et al. 2013). However, possibly the OPI1 deletion could improve d-glucaric acid production also in the pgi1-deficient strains by a still unknown mechanism. In addition, differences in the strain background, pathway genes, and culture conditions, e.g. aeration and medium composition, may contribute to the differences in D-glucaric acid amounts produced.
The S. cerevisiae pgi1-deficient strains do not tolerate d-glucose concentration above 2 g L−1, potentially decreasing their viability, and suitability to larger scale processes. Our fed-batch cultures did not drastically improve viability and more optimised feeding strategies would be needed. The viability was better on polymeric substrate α-cellulose where hydrolytic enzymes were used to release d-glucose, but the rate of d-glucose release would require further optimization to increase d-glucaric acid production. Recently, consolidated bioprocess (CBP) with concomitant sugar release and conversion to products has been developed for d-glucaric acid production (Fang et al. 2022, 2023; Li et al. 2021). This could be an option for the pgi1-deficient strains.
d-Glucaric acid production with S. cerevisiae has developed rapidly during recent years, especially considering volumetric titers (Table1). New myo-inositol oxygenases (Marques et al. 2020), and ways for improved viability (Guo et al. 2022) would be highly interesting to test in the pgi1-deficient S. cerevisiae. Also, uncoupling growth and production, or regulating the Pgi1p amount by synthetic promoters, gene switches, and/or degradation approaches like those implemented in E. coli (Brockman and Prather 2015; Gupta et al. 2017; Hou et al. 2020; Qu et al. 2018) could improve d-glucaric acid production and viability in the pgi1-deficient S. cerevisiae.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Abbott DA, Zelle RM, Pronk JT, van Maris AJ (2009) Metabolic engineering of Saccharomyces cerevisiae for production of carboxylic acids: current status and challenges. FEMS Yeast Res 9:1123–1136. https://doi.org/10.1111/j.1567-1364.2009.00537.x
Blank LM, Lehmbeck F, Sauer U (2005) Metabolic-flux and network analysis in fourteen hemiascomycetous yeasts. FEMS Yeast Res 5:545–558. https://doi.org/10.1016/j.femsyr.2004.09.008
Boer H, Maaheimo H, Koivula A, Penttilä M, Richard P (2010) Identification in Agrobacterium tumefaciens of the d-galacturonic acid dehydrogenase gene. Appl Microbiol Biotechnol 86:901–909. https://doi.org/10.1007/s00253-009-2333-9
Brockman IM, Prather KLJ (2015) Dynamic metabolic engineering: New strategies for developing responsive cell factories. Biotechnol J 10:1360–1369. https://doi.org/10.1002/biot.201400422
Cheah LC, Stark T, Adamson LSR, Abidin RS, Lau YH, Sainsbury F, Vickers CE (2021) Artificial self-assembling nanocompartment for organizing metabolic pathways in yeast. ACS Synth Biol 10:3251–3263. https://doi.org/10.1021/acssynbio.1c00045
Chen N, Wang J, Zhao Y, Deng Y (2018) Metabolic engineering of Saccharomyces cerevisiae for efficient production of glucaric acid at high titer. Microb Cell Fact 17:1–11. https://doi.org/10.1186/s12934-018-0914-y
Chen L-Z, Huang S-L, Hou J, Guo X-P, Wang F-S, Sheng J-Z (2020) Cell-based and cell-free biocatalysis for the production of d-glucaric acid. Biotechnol Biofuels 13:203. https://doi.org/10.1186/s13068-020-01847-0
Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P (1992) Multifunctional yeast high-copy-number shuttle vectors. Gene 110:119–122. https://doi.org/10.1016/0378-1119(92)90454-w
Ciriacy M, Breitenbach I (1979) Physiological effects of seven different blocks in glycolysis in Saccharomyces cerevisiae. J Bacteriol 139:152–160
de Koning W, van Dam K (1992) A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal Biochem 204:118–123
Doong SJ, Gupta A, Prather KLJ (2018) Layered dynamic regulation for improving metabolic pathway productivity in Escherichia coli. PNAS 115:2964–2969. https://doi.org/10.1073/pnas.1716920115
Dueber JE, Wu GC, Malmirchegini GR, Moon TS, Petzold CJ, Ullal AV, Prather KL, Keasling JD (2009) Synthetic protein scaffolds provide modular control over metabolic flux. Nat Biotechnol 27:753–759. https://doi.org/10.1038/nbt.1557
Fang H, Deng Y, Pan Y, Li C, Yu L (2022) Distributive and collaborative push-and-pull in an artificial microbial consortium for improved consolidated bioprocessing. AIChE J 68:e17844. https://doi.org/10.1002/AIC.17844
Fang H, Zhao C, Li C, Song Y, Yu L, Song X, Wu J, Yang L (2023) Direct consolidated bioprocessing for d-glucaric acid production from lignocellulose under subcritical water pretreatment. J Chem Eng 454:140339. https://doi.org/10.1016/J.CEJ.2022.140339
Fiaux J, Cakar ZP, Sonderegger M, Wuthrich K, Szyperski T, Sauer U (2003) Metabolic-flux profiling of the yeasts Saccharomyces cerevisiae and Pichia stipitis. Eukaryot Cell 2:170–180. https://doi.org/10.1128/ec.2.1.170-180.2003
Gancedo JM, Lagunas R (1973) Contribution of the pentose-phosphate pathway to glucose metabolism in Saccharomyces cerevisiae: a critical analysis on the use of labelled glucose. Plant Sci Lett 1:193–200
Gancedo C, Serrano R (1989) Energy yielding metabolism. In: Rose AH, Harrison JS (eds) The yeasts. Academic press, Cambridge, pp 205–259
Gietz RD, Sugino A (1988) New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74:527–534
Gueldener U, Heinisch J, Koehler GJ, Voss D, Hegemann JH (2002) A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30:e23. https://doi.org/10.1093/NAR/30.6.E23
Guo L, Qi M, Gao C, Ye C, Hu G, Song W, Wu J, Liu L, Chen X (2022) Engineering microbial cell viability for enhancing chemical production by second codon engineering. Metab Eng 73:235–246. https://doi.org/10.1016/J.YMBEN.2022.08.008
Gupta A, Hicks MA, Manchester SP, Prather KLJ (2016) Porting the synthetic d-glucaric acid pathway from Escherichia coli to Saccharomyces cerevisiae. Biotechnol J 11:1201–1208. https://doi.org/10.1002/biot.201500563
Gupta A, Reizman IMB, Reisch CR, Prather KLJ (2017) Dynamic regulation of metabolic flux in engineered bacteria using a pathway-independent quorum-sensing circuit. Nat Biotechnol 35:273–279. https://doi.org/10.1038/nbt.3796
Henry SA, Gaspar ML, Jesch SA (2014) The response to inositol: regulation of glycerolipid metabolism and stress response signaling in yeast. Chem Phys Lipids 180:23–43. https://doi.org/10.1016/J.CHEMPHYSLIP.2013.12.013
Heux S, Cadiere A, Dequin S (2008) Glucose utilization of strains lacking PGI1 and expressing a transhydrogenase suggests differences in the pentose phosphate capacity among Saccharomyces cerevisiae strains. FEMS Yeast Res 8:217–224. https://doi.org/10.1111/J.1567-1364.2007.00330.X
Hou J, Gao C, Guo L, Nielsen J, Ding Q, Tang W, Hu G, Chen X, Liu L (2020) Rewiring carbon flux in Escherichia coli using a bifunctional molecular switch. Metab Eng 61:47–57. https://doi.org/10.1016/J.YMBEN.2020.05.004
Lee CC, Kibblewhite RE, Paavola CD, Orts WJ, Wagschal K (2016) Production of glucaric acid from hemicellulose substrate by rosettasome enzyme assemblies. Mol Biotechnol 58:489–496. https://doi.org/10.1007/s12033-016-9945-y
Li C, Lin X, Ling X, Li S, Fang H (2021) Consolidated bioprocessing of lignocellulose for production of glucaric acid by an artificial microbial consortium. Biotechnol Biofuels 14:1–16. https://doi.org/10.1186/S13068-021-01961-7
Lien OG (1959) Determination of gluconolactone, galactonolactone and their free acids by the hydroxamate method. Anal Chem 31:1363–1366
Liu Y, Gong X, Wang C, Du G, Chen J, Kang Z (2016) Production of glucaric acid from myo-inositol in engineered Pichia pastoris. Enzyme Microb 91:8–16. https://doi.org/10.1016/j.enzmictec.2016.05.009
Maaheimo H, Fiaux J, Cakar ZP, Bailey JE, Sauer U, Szyperski T (2001) Central carbon metabolism of Saccharomyces cerevisiae explored by biosynthetic fractional 13C labeling of common amino acids. Eur J Biochem 268:2464–2479
Maitra PK (1971) Glucose and fructose metabolism in a phosphoglucoseisomeraseless mutant of Saccharomyces cerevisiae. J Bacteriol 107:759–769
Marques WL, Anderson LA, Sandoval L, Hicks MA, Prather KLJ (2020) Sequence-based bioprospecting of myo-inositol oxygenase (Miox) reveals new homologues that increase glucaric acid production in Saccharomyces cerevisiae. Enzyme Microb 140:109623. https://doi.org/10.1016/J.ENZMICTEC.2020.109623
Moon TS, Yoon SH, Lanza AM, Roy-Mayhew JD, Prather KL (2009) Production of glucaric acid from a synthetic pathway in recombinant Escherichia coli. AEM 75:589–595. https://doi.org/10.1128/AEM.00973-08
Moon TS, Dueber JE, Shiue E, Prather KL (2010) Use of modular, synthetic scaffolds for improved production of glucaric acid in engineered E. coli. Metab Eng 12:298–305. https://doi.org/10.1016/j.ymben.2010.01.003
Nidelet T, Brial P, Camarasa C, Dequin S (2016) Diversity of flux distribution in central carbon metabolism of S. cerevisiae strains from diverse environments. Microb Cell Fact 15:1–13. https://doi.org/10.1186/S12934-016-0456-0
Nygård Y, Toivari MH, Penttilä M, Ruohonen L, Wiebe MG (2011) Bioconversion of d-xylose to d-xylonate with Kluyveromyces lactis. Metab Eng 13:383–391. https://doi.org/10.1016/j.ymben.2011.04.001
Petroll K, Care A, Bergquist PL, Sunna A (2020) A novel framework for the cell-free enzymatic production of glucaric acid. Metab Eng 57:162–173. https://doi.org/10.1016/j.ymben.2019.11.003
Qu YN, Yan HJ, Guo Q, Li JL, Ruan YC, Yue XZ, Zheng WX, Tan TW, Fan LH (2018) Biosynthesis of d-glucaric acid from sucrose with routed carbon distribution in metabolically engineered Escherichia coli. Metab Eng 47:393–400. https://doi.org/10.1016/j.ymben.2018.04.020
Raman S, Rogers JK, Taylor ND, Church GM (2014) Evolution-guided optimization of biosynthetic pathways. PNAS 111:17803–17808. https://doi.org/10.1073/pnas.1409523111
Reizman IMB, Stenger AR, Reisch CR, Gupta A, Connors NC, Prather KLJ (2015) Improvement of glucaric acid production in E. coli via dynamic control of metabolic fluxes. Metab Eng Commun 2:109–116. https://doi.org/10.1016/J.METENO.2015.09.002
Rogers JK, Church GM (2016) Genetically encoded sensors enable real-time observation of metabolite production. PNAS 113:2388–2393. https://doi.org/10.1073/pnas.1600375113
Sakuta R, Nakamura N (2019) Production of hexaric acids from biomass. International J Mol Sci 20:3660. https://doi.org/10.3390/IJMS20153660
Salusjärvi L, Toivari M, Vehkomäki M-L, Koivistoinen O, Mojzita D, Niemelä K, Penttilä M, Ruohonen L (2017) Production of ethylene glycol or glycolic acid from d-xylose in Saccharomyces cerevisiae. Appl Microbiol Biotechnol 101:8151–8163. https://doi.org/10.1007/s00253-017-8547-3
Sherman F, Fink G, Hicks JB (1983) Methods in yeast genetics: A laboratory manual. Cold Springs Harbor Laboratory, Cold Springs Harbor
Shiue E, Prather KLJ (2014) Improving d-glucaric acid production from myo-inositol in E. coli by increasing MIOX stability and myo-inositol transport. Metab Eng 22:22–31. https://doi.org/10.1016/j.ymben.2013.12.002
Shiue E, Brockman IM, Prather KLJ (2015) Improving product yields on d-glucose in Escherichia coli via knockout of pgi and zwf and feeding of supplemental carbon sources. Biotechnol Bioeng 112:579–587. https://doi.org/10.1002/BIT.25470
Su HH, Guo ZW, Wu XL, Xu P, Li N, Zong MH, Lou WY (2019) Efficient bioconversion of sucrose to high-value-added glucaric acid by in vitro metabolic engineering. Chemsuschem 12:2278–2285. https://doi.org/10.1002/cssc.201900185
Su HH, Peng F, Ou XY, Zeng YJ, Zong MH, Lou WY (2020) Combinatorial synthetic pathway fine-tuning and cofactor regeneration for metabolic engineering of Escherichia coli significantly improve production of d-glucaric acid. N Biotechnol 59:51–58. https://doi.org/10.1016/j.nbt.2020.03.004
Toivari MH, Maaheimo H, Penttilä M, Ruohonen L (2010a) Enhancing the flux of d-glucose to the pentose phosphate pathway in Saccharomyces cerevisiae for the production of d-ribose and ribitol. Appl Microbiol Biotechnol 85:731–739. https://doi.org/10.1007/s00253-009-2184-4
Toivari MH, Ruohonen L, Richard P, Penttilä M, Wiebe MG (2010b) Saccharomyces cerevisiae engineered to produce d-xylonate. Appl Microbiol Biotechnol 88:751–760. https://doi.org/10.1007/s00253-010-2787-9
van Strien N, Rautiainen S, Asikainen M, Thomas DA, Linnekoski J, Niemelä K, Harlin A (2020) A unique pathway to platform chemicals: aldaric acids as stable intermediates for the synthesis of furandicarboxylic acid esters. Green Chem 22:8271–8277. https://doi.org/10.1039/d0gc02293d
Verho R, Richard P, Jonson PH, Sundqvist L, Londesborough J, Penttilä M (2002) Identification of the first fungal NADPH-GAPDH from Kluyveromyces lactis. Biochem 41:13833–13838. https://doi.org/10.1021/bi0265325
Verma BK, Mannan AA, Zhang F, Oyarzún DA (2022) Trade-offs in biosensor optimization for dynamic pathway engineering. ACS Synth Biol 11:228–240. https://doi.org/10.1021/ACSSYNBIO.1C00391
Vinopal RT, Hillman JD, Schulman H, Reznikoff WS, Fraenkel DG (1975) New phosphoglucose isomerase mutants of Escherichia coli. J Bacteriol 122:1172–1174
Ye C, Bandara WMMS, Greenberg ML (2013) Regulation of inositol metabolism is fine-tuned by inositol pyrophosphates in Saccharomyces cerevisiae. JBC 288:24898–24908. https://doi.org/10.1074/jbc.M113.493353
Yoon SH, Moon TS, Iranpour P, Lanza AM, Prather KJ (2009) Cloning and characterization of uronate dehydrogenases from two pseudomonads and Agrobacterium tumefaciens strain C58. J Bacteriol 191:1565–1573. https://doi.org/10.1128/jb.00586-08
Zhang X, Xu C, Liu YL, Wang J, Zhao YY, Deng Y (2020) Enhancement of glucaric acid production in Saccharomyces cerevisiae by expressing Vitreoscilla hemoglobin. Biotechnol Lett 42:2169–2178. https://doi.org/10.1007/s10529-020-02966-2
Zhang Q, Wan Z, Yu IKM, Tsang DCW (2021) Sustainable production of high-value gluconic acid and glucaric acid through oxidation of biomass-derived glucose: a critical review. J Clean Prod 312:127745. https://doi.org/10.1016/J.JCLEPRO.2021.127745
Zhao Y, Li J, Su R, Liu Y, Wang J, Deng Y (2021) Effect of magnesium ions on glucaric acid production in the engineered Saccharomyces cerevisiae. J Biotechnol 332:61–71. https://doi.org/10.1016/J.JBIOTEC.2021.03.020
Zhao Y, Zuo F, Shu Q, Yang X, Deng Y (2023) Efficient production of glucaric acid by engineered Saccharomyces cerevisiae. AEM. https://doi.org/10.1128/aem.00535-23
Acknowledgements
This study was financially supported by the Academy of Finland through the grants Centre of Excellence in White Biotechnology—Green Chemistry (grant 118573), and SA SBio (grant 310191), and by the European Commission through the Seventh Framework Programme (FP7/2007-2013) under grant agreement N° FP7-241566 BIOCORE which are gratefully acknowledged. We thank Toni Paasikallio for assistance with fermentations, and Tuulikki Seppänen-Laakso, Kaarina Viljanen and Matti Hölttä for assistance with GC-MS measurements.
Funding
Open Access funding provided by Technical Research Centre of Finland. This study was financially supported by the Academy of Finland through the grants Centre of Excellence in White Biotechnology—Green Chemistry (grant 118573), and SA SBio (grant 310191), and by the European Commission through the Seventh Framework Programme (FP7/2007-2013) under grant agreement N° FP7-241566 BIOCORE.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Strain engineering, shake flask cultures, and HPLC analyses were performed by MT, and M-LV. Bioreactor cultures, HPLC analyses, viability, and lactone assay were conducted and supervised by MGW. MT and MGW drafted the manuscript. All authors participated in result analysis, and commented previous manuscript versions, and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
This article does not contain any studies with human or animal subjects.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Toivari, M., Vehkomäki, ML., Ruohonen, L. et al. Production of d-glucaric acid with phosphoglucose isomerase-deficient Saccharomyces cerevisiae. Biotechnol Lett 46, 69–83 (2024). https://doi.org/10.1007/s10529-023-03443-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10529-023-03443-2